
Organotin(iv) carboxylate derivatives as a new addition to anticancer and antileishmanial agents: design, physicochemical characterization and interaction with Salmon sperm DNA†
Abstract
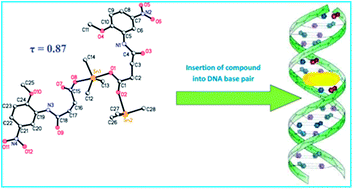
This article demonstrates the synthesis and characterization of novel organotin(IV) carboxylate complexes and their medicinal applications. Metal complexes and organometallic compounds have growing importance in medicine, particularly in oncology. Organotin derivatives have caught much attention during the last two decades for their potential biocidal activities. In recent years several organotin compounds have been synthesized, some with interesting cytotoxic properties. Here we will focus on the relevance of organotin derivatives as very promising potential candidates in anticancer therapy. Ten new organotin(IV) complexes were synthesized and characterized by using FT-IR, NMR (1H, 13C and 119Sn), elemental analysis, mass spectrometry and single crystal X-ray techniques. The results of infrared spectroscopy of the sodium salt and complexes showed that the coordination took place through the oxygen atoms of the carboxylate group. Crystallographic data for complexes 1 and 3 showed that the tin has a distorted trigonal bipyramidal geometry with the three alkyl groups in the trigonal plane and the two oxygen atoms in the equatorial plane. In the case of complex 4 the geometry is tetrahedral due to the steric hindrance of the bulky phenyl groups. The compounds were screened for in vitro antiproliferative activity against lung carcinoma (H-157) and kidney fibroblast (BHK-21) cell lines and revealed significant anticancer activity. They were also tested for antileishmanial activity against the promastigote form of leishmania major and exhibited IC50 value 0.98 ± 0.06 μM (Compound 10) as compared to amphotericin B (IC50 value 0.29 ± 0.05 μM). The synthesized compounds act as a potent intercalator of SS-DNA and insert themselves into the nitrogenous bases of the DNA resulting in hypochromism and bathochromic shift.
 Please wait while we load your content...
Please wait while we load your content...

