
Controllable synthesis of biosourced blue-green fluorescent carbon dots from camphor for the detection of heavy metal ions in water†
Abstract
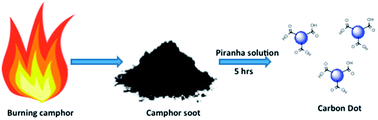
A robust method for the synthesis of fluorescent carbon dots (C dots) from camphor, which provides an insight into the mechanism of C dot formation, is reported. Camphor is a biosourced hydrocarbon, which contains an hexagonal ring arranged like an open book. Burning of camphor leads to the formation of soot, which comprises graphitic domains. The soot when treated with piranha solution disintegrates into smaller domains leading to the formation of C dots with a size distribution of ∼1–4 nm. The C dots obtained were carboxyl terminated which was confirmed from the infrared spectroscopic measurements. The D and G bands at ∼1314 cm−1 and ∼1586 cm−1, respectively, found using Raman spectroscopy and the peaks at 25.01° found using X-ray diffraction of C dots confirm the presence of graphitic domains. Photoluminescence studies were carried out which reveal exceptional fluorescence in the as prepared C dots. Interestingly the quantum yield is found to be around 21.16%, which is significantly higher than the values reported in previous papers. The current study deals with the sensing of metal ions. Heavy metal cations such as Cd2+ and Hg2+ were used to check whether they affect the fluorescence properties of C dots. It was found that other metal ions like Cu2+, Fe2+ and Zn2+ also quenched the fluorescence of C dot with a different quenching profile.
 Please wait while we load your content...
Please wait while we load your content...

