DOI:
10.1039/C4RA10304A
(Paper)
RSC Adv., 2014,
4, 53921-53926
Evaluation of efficiencies of radiolysis, photocatalysis and ozonolysis of modified simulated textile dye waste-water
Received
12th September 2014
, Accepted 25th September 2014
First published on 26th September 2014
Abstract
The efficiencies of the advanced oxidation processes (AOPs), viz. electron beam radiolysis in the presence of K2S2O8, gamma radiolysis in the presence of K2S2O8, photocatalysis, photocatalysis in the presence of K2S2O8, photolysis in presence of K2S2O8, ozonolysis and ozonolysis in the presence of K2S2O8, were systematically investigated for the treatment of modified (pH adjusted with H2SO4) simulated textile dye waste-water (MSTDWW) containing Reactive Red-120. The efficiencies of these AOPs were investigated in terms of their oxygen-equivalent chemical oxidation capacity, and the cost of energy and other ancillary inputs. The least amount of oxidant was required by electron beam radiolysis compared to the other AOPs studied here, to achieve the same extent of mineralization of MSTDWW. To the best of our knowledge, this study is the first to report an approach to calculate the equivalent cost of gamma radiolysis in comparison to other AOPs consuming electrical energy. Among these AOPs, the electron beam treatment of MSTDWW in the presence of K2S2O8 had the lowest effective cost.
1. Introduction
Textile effluents are hazardous to the ecosystem, because of the implementation of intense colour to water from the bio-resistant synthetic dye molecules even at very low concentrations. Moreover, the auxiliary chemicals (such as surfactants, sequestering agents, pH-adjusting acids, and inorganic salts) of the dye bath contribute to ∼83% of the organic load of the effluent.1,2 The heavy organic load of the textile effluent causes a negative impact to aquatic lives, owing to the decrease in the dissolved oxygen concentration in the water stream.3 The chemical oxygen demand (COD), which is a measure of the organic load of the effluent, varies in the range of 2900–3000 ppm in textile effluents. Since this is well above the permissible discharge limit (COD ≤ 250 ppm) set by the Central Pollution Control Board under the Ministry of Environment and Forest, Government of India, the development of an effective and efficient treatment process for textile effluents is highly necessary.
Synthetic reactive azo dyes are mostly used in textile industries, although they are typically hydrolyzed in an alkaline dye bath solution.4 Physico-chemical treatments, such as precipitation, coagulation, reverse osmosis, electrolysis and adsorption are used in conventional dye effluent treatment plants.5 However, the large amount of sludge or secondary waste generation, as well as the resistance of the reactive dyes to photo- or bio- or chemical-degradation are the major disadvantages of physico-chemical treatment processes.6
The heavy organic loads of the textile effluents come from the auxiliary organic components, such as sodium dodecylbenzenesulfonate (SDBS, detergent used for washing out the excess dye), ethylenediaminetetraacetic acid (EDTA, for removing unwanted metal ions from the dye bath), acetic acid (CH3COOH, for adjusting the pH of the dye bath to 10) used in the dye bath. On replacing the organic acid (CH3COOH) by an inorganic acid (H2SO4) in the pH adjustment step, we could bring down the COD of the simulated textile dye waste-water to 245 ppm by gamma radiolysis at 60 kGy in the presence of potassium persulfate (K2S2O8).7 A comparative study on the process efficiencies of three major advanced oxidation processes (AOPs) viz. radiolysis (gamma and electron beam (EB)), photocatalysis, and ozonolysis for the treatment of modified (pH adjusted with H2SO4) simulated textile dye waste-water (MSTDWW) of Reactive Red-120 (RR-120) was investigated in terms of the oxygen-equivalent chemical oxidation capacity (OCC) and the cost of energy and other ancillary inputs.
2. Experimental
2.1 Dye and chemicals
RR-120, SDBS, EDTA, K2S2O8 and tert-butanol were purchased from Sigma-Aldrich. Titanium dioxide (TiO2) (Degussa (P25), particle size ∼30 nm) was used as the photocatalyst. All other chemicals used were of the highest purity and were used without any pretreatment.
2.2 Preparation of MSTDWW
The dye bath composition was kept fixed, as reported by Paul Guin et al., except for the neutralizing acid.7 The hydrolyzed dye effluent was prepared by refluxing the constituents with 1 M NaOH at 80–90 °C for 3 hours;8,9 followed by the adjustment of the pH to 10 by H2SO4. This solution will henceforth be termed as MSTDWW.
2.3 Instruments and analytical procedures
The steady state radiolysis of MSTDWW solution was carried out using 60Co gamma radiation in a gamma chamber GC-5000 with a dose rate of 1.3 kGy h−1, as determined using Fricke dosimetry [G(Fe3+) = 15.6/100 eV−1]. EB irradiation was performed with a 10 MeV pulsed accelerator at a beam current of 33 mA and a 10 kGy dose/pass. The dosimetry for EB was performed using a FWT60 radio-chromic film dosimeter calibrated with a graphite calorimeter. The photocatalytic experiments were carried out using Rayonet Photochemical Reactor containing 16 mercury lamps of 8 W power each. The lamps emit photons in the near-UV region (mainly around 350 nm) with a flux of 5.0 × 1015 photons cm−2 s−1. Ozone was generated from pure oxygen using an ozone generator (Model UOS02). The rate of flow of ozone through the solution was determined as 6.7 × 10−3 mol dm−3 min−1 by the iodometric method. The COD of the solutions was measured using a Spectroquant® Pharo 300 COD analyzer.
3. Results and discussion
3.1 Radiolysis of MSTDWW in the presence of K2S2O8
The radiolysis of water in the pH range of 3–11 produces three main reactive species, viz. hydrated electron (eaq−), ˙OH radical and hydrogen atom (H˙). The G-values [species per 100 eV] of the intermediates are given below.10| | |
G(eaq−) = 2.7; G(˙OH) = 2.7; G(H˙) = 0.6
| (1) |
During the radiolysis of water in the presence of K2S2O8, the eaq− and H˙ preferentially react with S2O82− producing SO4˙− (Reduction potential = 2.5–3.1 V vs. NHE), which can subsequently react with different components of MSTDWW present in the system.
| | |
eaq− + S2O82− → SO4˙− + SO42−
| (2) |
| | |
S2O82− + H˙ → SO4˙− + HSO4−
| (3) |
In addition, ˙OH radicals (Reduction potential = 2.7 V vs. NHE) can also react independently with those components of MSTDWW. Gamma radiolysis in the presence of K2S2O8 has recently been proven to be an efficient technique for the mineralization of the organic molecules.7,11 The higher extent of mineralization of organic molecules during radiolysis in the presence of K2S2O8 compared to the radiolysis in the absence of K2S2O8 happens because of the preferential formation of benzyl type radicals via the formation of benzene radical cations. Therefore, MSTDWW was irradiated for various doses at pH 10.0 in the presence of 4 × 10−2 mol dm−3 K2S2O8. The mineralization extent at different doses is shown in Fig. 1a. It shows 20% and 75% mineralizations at doses of 11 kGy and 60 kGy, respectively. It could be noted that, at the same time, only 16% and 54% mineralizations were observed for the STDWW (pH adjusted with CH3COOH).7 Furthermore, ∼80% mineralization was observed for the gamma radiolysis of MSTDWW at the 60 kGy dose, while only 60% mineralization was observed for the gamma radiolysis of STDWW at 100 kGy.7 Therefore, the nature of the pH adjusting acid influences the extent of the mineralization of MSTDWW with that of the STDWW. It is important to mention that K2S2O8 itself can produce SO4˙− by thermal decomposition at 38–40°C, which is the usual temperature of the solution during gamma radiolysis.12 Therefore, the extent of mineralization of MSTDWW in the presence of 4 × 10−2 mol dm−3 K2S2O8 was studied at 40 °C under room conditions (no irradiation), and here, no appreciable mineralization of MSTDWW was observed.
 |
| | Fig. 1 Mineralization of MSTDWW in the presence of 4 × 10−2 mol dm−3 K2S2O8 during (a) gamma radiolysis, and (b) EB radiolysis. | |
The MSTDWW solution was irradiated at different doses by EB at pH 10.0 in the presence of 4 × 10−2 mol dm−3 K2S2O8. The extent of mineralization of MSTDWW increased with each dose by ∼20% (Fig. 1b). The high intensity electron beam rapidly deposits energy to the aqueous solution and elevates the temperature of the aqueous solution from ambient temperature.13,14 Therefore, it can be speculated that the high yield of ˙OH radical and SO4˙− (by the conjugated effects of radiolytic and thermal decompositions) enhances the %-mineralization of MSTDWW to ∼34% and 96% at 11 kGy and 60 kGy doses, respectively. At 60 kGy, the COD of the final solution was brought down to below 100 ppm, which is below the recommended limit of discharge (≤250 ppm).
3.2 Photocatalysis of MSTDWW
On illuminating the photocatalyst, such as TiO2, with UV light, the electrons from the valence bands (VB) are promoted to the conduction bands (CB), generating a hole in the VB. The promoted electron in the CB reacts with the dissolved oxygen, producing O2˙− and HO2˙, whereas, the hole generated in the VB can react with either with the organic moiety or OH−. The reaction of the hole with OH− produces a ˙OH radical, which can also oxidize the organic moieties.9 The complete photocatalytic cycle is shown in reactions (4)–(9):| | |
TiO2 + hν(UV) → TiO2(eCB− + hVB+)
| (4) |
| | |
TiO2(hVB+) + H2O → TiO2 + H+ + ˙OH
| (5) |
| | |
TiO2(hVB+) + OH− → TiO2 + ˙OH
| (6) |
| | |
TiO2(eCB−) + O2 → TiO2 + O2˙−
| (7) |
| | |
Organics + ˙OH → Degradation products
| (9) |
where hν is the photon energy required to excite the semiconductor electron from the VB to CB.
The photocatalysis of MSTDWW was carried out over different time intervals. However, only 30% mineralization of MSTDWW was observed after 10 hours of photocatalytic treatment (Fig. 2a), and no appreciable change in the extent of mineralization was observed over longer times. It could be noted that only 24% mineralization of STDWW was observed under the same photocatalytic conditions.7 It is important to mention that the TiO2 particles made very stable suspensions with the aqueous solution of the individual components of MSTDWW. However, this settled down rapidly in MSTDWW. We speculated that the presence of high salt concentrations (∼2 × 104 ppm of NaCl) in the MSTDWW might be responsible for changing the surface properties of the TiO2 particles, which finally leads to the easy settlement of the catalyst in MSTDWW. Since, we did not have a photoreactor with stirring facilities, we can only speculate on this, and we could not confirm that the rapid settlement of the catalyst in MSTDWW might be one of the reasons behind the poor efficiency of the photocatalytic mineralization of MSTDWW. On the other hand, the coulombic repulsion between the negatively charged surface of TiO2 (pHpzc = 6.0 ± 0.2) and OH− (at pH 10.0) could also prevent the production of ˙OH, resulting in the poor mineralization of MSTDWW.7,9,15
 |
| | Fig. 2 Mineralization of MSTDWW over different durations in: (a) photocatalysis, (b) photocatalysis in the presence of 4 × 10−2 mol dm−3 K2S2O8, (c) photolysis in presence of 4 × 10−2 mol dm−3 K2S2O8, (d) ozonolysis, and (e) ozonolysis in the presence of 4 × 10−2 mol dm−3 K2S2O8. | |
Instead of molecular oxygen, S2O82− can also take the CB electron from TiO2 nanoparticles, thereby producing SO4˙− radicals (reaction (10)):16
| | |
S2O82− + eCB− → 2SO4˙−
| (10) |
Therefore, the effect of K2S2O8 on the photocatalytic degradation of MSTDWW was also investigated in the presence of 4 × 10−2 mol dm−3 K2S2O8. The extent of mineralization increased by ∼10–12% during the photocatalysis of MSTDWW in the presence of K2S2O8 (Fig. 2b). The increase in the %-mineralization of MSTDWW during the photocatalysis in the presence of K2S2O8 is attributed to: (i) the decrease in the probability of recombination of the photogenerated electrons and holes, and (ii) the formed SO4˙− having a higher mineralization efficiency.7,16 However, the application of this process is limited by the coulombic repulsion between the negatively charged surface of TiO2 (pHpzc = 6.0 ± 0.2) and S2O82− (at pH 10.0) and the rapid settlement of the catalyst in MSTDWW.
The photolysis (photochemical decomposition of K2S2O8 in the absence of TiO2) did not impart any enhancement in the extent of mineralization of MSTDWW (Fig. 2c). This is speculated by the lower yield of SO4˙− radicals from the photolysis of S2O82− with ∼350 nm UV light.16
3.3 Ozonolysis of MSTDWW
Ozone, which itself is a strong oxidant (Reduction potential = 2.07 V vs NHE), can produce more powerful oxidants, i.e. ˙OH, under alkaline conditions (pH 10.0) through reactions (11)–(13).17,18| | |
O3 + H2O → 2˙OH + O2
| (11) |
| | |
O3 + OH− → O2˙− + HO2˙
| (12) |
| | |
O3 + HO2˙ → 2O2 + ˙OH
| (13) |
Therefore, ˙OH is assumed to be the predominant reactive species available under the experimental conditions.19,20 The extent of mineralization of MSTDWW was studied at different durations of ozonolysis at pH 10.0. About 30% and 60% extent of mineralizations of MSTDWW were achieved after 0.5 hours and 4 hours of ozonolysis, respectively. However, after 4 hours, no significant increase in the extent of mineralization was observed (Fig. 2d). It should be noted that only 13% and 25% mineralization of STDWW was observed under the same ozonolytic conditions.7 The effect of K2S2O8 on the ozonolysis of MSTDWW was investigated in the presence of 4 × 10−2 mol dm−3 K2S2O8 (Fig. 2e). The extent of mineralization decreased drastically in case of ozonolysis in the presence of K2S2O8. The K2S2O8 does not produce SO4˙− during ozonolysis (in the absence of any radiation or thermal activation); instead, some of the ˙OH radicals formed during ozonolysis will react with K2S2O8 giving some products (reaction (14)) which may not react with components of MSTDWW.
| | |
˙OH + K2S2O8 → products
| (14) |
Therefore, the extent of %mineralization decreased during ozonolysis in presence of K2S2O8.
3.4 Comparison of the process efficiencies of radiolysis, photocatalysis, and ozonolysis for the mineralization of MSTDWW
3.4.1. In terms of the OCC. The process efficiencies of different AOPs were compared in terms of OCC, which is defined as the kg of O2 equivalent to the quantity of oxidant reagents used in an AOP to treat 1 m3 of waste-water.21 This gives an index of the chemical efficiency of the oxidants used in an AOP by quantifying the amount of the oxidants (kg O2) added per m3 of waste-water. The OCC of radiolysis (gamma and EB) (eqn (15)), photocatalysis/photolysis (eqn (16)), and ozonolysis (eqn (17)) are calculated by the following eqn (15–17):7| |
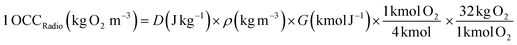 | (15) |
where, D is the dose, ρ is the density of water, G (SO4˙−) = 3.4 × 10−10 kmol J−1 or 3.3/100 eV; G (˙OH) = 2.8 × 10−10 kmol J−1 or 2.7/100 eV.| |
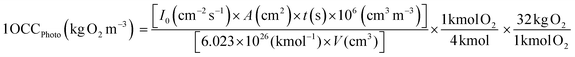 | (16) |
| |
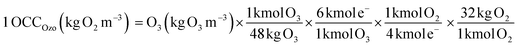 | (17) |
Fig. 3 shows that the lowest degree of mineralization of MSTDWW (to an extent of 28%) was observed in the photocatalysis and photolysis of K2S2O8. Thus OCC values and the cost of the energy source and other ancillary inputs of different AOPs were compared only for 28% mineralization of MSTDWW. It could be noted that the OCCs of radiolysis, photocatalysis, ozonolysis, and radiolysis (+K2S2O8) of STDWW could be calculated only for 16% mineralization, which was the lowest observed degree of mineralization of STDWW.7 The OCC values of photocatalysis, photocatalysis (+K2S2O8), photolysis in presence of K2S2O8, ozonolysis, ozonolysis (+K2S2O8), and gamma (+K2S2O8) and EB (+K2S2O8) radiolysis for 28% mineralization were calculated to be 6.29, 2.46, 7.63, 9.29, 38.83, 0.08, and 0.04 kg equivalent O2 m−3, respectively. EB radiolysis in the presence of K2S2O8 showed a maximum chemical efficiency (∼96% mineralization) of the oxidants at 0.3 kg equivalent of O2 m−3 OCC. About 78% mineralization was observed in gamma radiolysis, and less than 10% mineralizations were observed for others at 0.3 kg equivalent of O2 m−3 OCC. It could be noted that 0.3 kg equivalent of O2 m−3 OCC could mineralize only 54% of STDWW by gamma radiolysis in the presence of K2S2O8.7 Therefore, it could be safely concluded that the amount of oxidant required to achieve the same extent of mineralization of MSTDWW by EB radiolysis was the least, compared to other processes studied here. Therefore, the OCC for a 28% mineralization of MSTDWW follows the order: EB (+K2S2O8) radiolysis < gamma (+K2S2O8) radiolysis < photocatalysis (+K2S2O8) < photocatalysis ∼ photolysis in presence of K2S2O8 < ozonolysis < ozonolysis (+K2S2O8). The mechanism of enhancement in the extent of mineralization of STDWW (pH adjusted by acetic acid) and ibuprofen during radiolysis in the presence of K2S2O8 has been studied in depth by the authors.7,11 Since, only the nature of the pH adjusting acid changes, we speculate that the mechanism of mineralization of the components of MSTDWW was quite similar to that of STDWW.7,11
 |
| | Fig. 3 Mineralization of MSTDWW with OCC for (a) photocatalysis, (b) photocatalysis in the presence of 4 × 10−2 mol dm−3 K2S2O8, (c) photolysis in presence of 4 × 10−2 mol dm−3 K2S2O8, (d) ozonolysis, (e) ozonolysis in the presence of 4 × 10−2 mol dm−3 K2S2O8, (f) gamma radiolysis in the presence of 4 × 10−2 mol dm−3 K2S2O8 and (g) EB radiolysis in the presence of 4 × 10−2 mol dm−3 K2S2O8. | |
3.4.2. In terms of the cost of the energy source and other ancillary inputs. The efficiencies of EB (+K2S2O8) and gamma (+K2S2O8) radiolysis, photocatalysis (+K2S2O8), and ozonolysis were evaluated in terms of the cost of energy and other ancillary inputs. The cost of the electrical energy required for EB (+K2S2O8) radiolysis, photocatalysis (+K2S2O8), and ozonolysis can be calculated using eqn (18).| | |
EEC = P × (t/60) × (1000/v)
| (18) |
where EEC (in kWh/m3) is the electric energy consumed (in kWh) to degrade a contaminant in unit volume (in m3), P is the rated power (in kW) of the AOP system, t is the duration (in min) of treatment, and v is the volume (in L) of MSTDWW treated in time t.The duration of treatment for 28% mineralization of MSTDWW by EB (+K2S2O8) radiolysis, photocatalysis (+K2S2O8), and ozonolysis were observed to be 0.6, 180, and 30 min, respectively. The cost of the electrical energy, along with the ancillary chemicals (if any), for these AOPs are summarized in Table 1. Among these processes, the costs involved in EB (+K2S2O8) treatment were the lowest.
Table 1 Comparison of the cost of energy and ancillary chemicals for different AOPs
| |
Ozonolysis |
Photocatalysis (+K2S2O8) |
EB radiolysis (+K2S2O8) |
| Guided by the maximum volume capacity of the instrument to treat the MSTDWW under the same treatment condition. |
| Power employed in the process (kW) |
0.08 |
0.128 |
1 |
| Treatment time (min) |
30 |
180 |
0.6 |
| Volume of MSTDWW treated (L)a |
0.04 |
0.26 |
1.9 |
| EEC (kWh)/m3 of MSTDWW |
1000 |
1477 |
5.3 |
| Electrical energy cost (INR)/m3 of MSTDWW @INR 8.5/kWh |
8500 |
12 554 |
45 |
| Cost of additional chemicals/gas (INR)/m3 of MSTDWW |
246![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (O2 cylinder cost @ INR 164/m3) 000 (O2 cylinder cost @ INR 164/m3) |
3000 (TiO2 cost @ INR 3/g) + 12975 (K2S2O8 cost @ INR 1.2/g) |
12975 (K2S2O8 cost @ INR 1.2/g) |
| Total cost/m3 of MSTDWW |
254500 |
28529 |
13020 |
In the gamma radiolysis of MSTDWW (which did not involve electrical energy), the cost of the energy source could be estimated by accounting for five effective half-lives of 60Co source using the following eqn (19)
| | |
CTP = I × R × t/(t1/2 × 5 × 365 × 24) × (1000/v)
| (19) |
where CTP (INR/m
3) is the average cost of the treatment process in Indian rupee (INR),
I is the initial activity in Curie (Ci) of the
60Co source,
R is the price (in INR) of
60Co source/Ci,
t is the treatment time (in hours),
t1/2 is the half-life (hour) of
60Co, and
v is the maximum volume capacity (in L) of the gamma chamber that can be treated in time
t. In our study, the initial activity of
60Co was 10
000 Ci, involving a cost of INR 70/Ci, and volume of the gamma chamber was 5 L. Therefore, the cost for 28% mineralization using gamma radiolysis was calculated as INR 7931/m
3. The total cost of gamma radiolysis (+K
2S
2O
8) for the treatment of MSTDWW was found to be INR 20906/m
3. This is the first approach to calculate the equivalent cost of gamma radiolysis in comparison to other AOPs consuming electrical energy.
The above results showed that the cost involved in EB (+K2S2O8) treatment was the lowest one among the studied AOPs for the mineralization of MSTDWW. It is important to note that the AOPs are emerging technologies currently being commercialized worldwide. A few UV/H2O2-based AOPs have been internationally commercialized for the treatment of drinking water and industrial water, using the advantage of both chemical and energy inputs.22–24 There are a few companies, such as AST clean water technologies, China; Trojan Technologies, Canada; Calgon Carbon Corporation and Xylem Global, US, who have brought some of the AOPs to international markets. In parallel, the radiation technology is internationally emerging for waste-water treatment.25–27 Radiation-based pilot sludge treatment plants have been established in New Mexico, USA (Gamma); Weldel, Germany (EB); Verginia Key, USA (EB); Takasaki, Japan (EB); Sao Paulo, Brazil (EB); Tucuman, Argentina (Gamma); and Daejeon, Korea (EB).25–27 In addition, radiation-based commercial sludge treatment plants have also been established in Vadodara, India (Gamma), and Munich, Germany.25–27 A pilot plant for treating 1000 m3 day−1 of dyeing waste-water with EB has been constructed and operated since 1998 in Daegu, Korea, together with a biological treatment facility.28,29 Therefore, we understand that the studies presented in this paper have a lot of scope to advance radiation-based technologies for the treatment of textile effluents. Furthermore, the EB has the ability to simultaneously disinfect the water during the degradation process.30 At this stage, the used EB (+K2S2O8) treatment process does not produce water suitable for reuse or for drinking. Hence, a multi-step treatment system would need to be designed in the near future.
4. Conclusions
This study explores a reliable, promising, and cost effective way to use EB radiolysis in the presence of K2S2O8 for the complete mineralization of recalcitrant organics in to CO2 and H2O. The least amount of oxidant was required for EB radiolysis in the presence of K2S2O8 to achieve the same extent of mineralization, compared to gamma radiolysis in the presence of K2S2O8, photocatalysis, photocatalysis in the presence of K2S2O8, photolysis in presence of K2S2O8, ozonolysis, and ozonolysis in the presence of K2S2O8. To the best of our knowledge, this study is the first to report an approach to calculate the equivalent cost of gamma radiolysis in comparison to other AOPs consuming electrical energy. Among these processes, the costs involved in EB treatment in the presence of K2S2O8 were the lowest.
Acknowledgements
The authors wish to sincerely thank Dr (Smt) S. Dhanya, RPCD, BARC, for her kind support during the work and Mr Mukesh Kumar, APPD, BARC, for his kind support in carrying out electron beam irradiation.
Notes and references
- I. Arslan, J. Hazard. Mater., 2001, 85, 229–241 CrossRef CAS PubMed.
- I. Arslan-Alanton and I. Alaton, Ecotoxicol. Environ. Saf., 2007, 68, 98–107 CrossRef PubMed.
- R. O. Cristovao, A. P. M. Tavares, J. M. Loureiro, R. A. R. Boaventura and E. A. Macedo, Bioresour. Technol., 2009, 100, 6236–6242 CrossRef CAS PubMed.
- J. Riu, I. Schonsee and D. Barcelo, Trends Anal. Chem., 1997, 16, 405–419 CrossRef CAS.
- D. Georgiou, A. Aivazidis, J. Hatiras and K. Gimouhopoulas, Water Res., 2003, 37, 2248–2250 CrossRef CAS PubMed.
- A. Pandey, P. Singh and L. Iyengar, Int. Biodeterior. Biodegrad., 2007, 59, 73–84 CrossRef CAS.
- J. Paul Guin, D. B. Naik, Y. K. Bhardwaj and L. Varshney, RSC Adv., 2014, 4, 39941–39947 RSC.
- M. Koch, A. Yediler, D. Lienert, G. Insel and A. Ketrupp, Chemosphere, 2002, 46, 109–113 CrossRef CAS PubMed.
- U. G. Akapon and B. H. Hameed, J. Hazard. Mater., 2009, 170, 520–529 CrossRef PubMed.
- N. Getoff, Radiat. Phys. Chem., 2002, 65, 437–446 CrossRef CAS.
- J. Paul (Guin), D. B. Naik, Y. K. Bhardwaj and L. Varshney, Radiat. Phys. Chem., 2014, 100, 38–44 CrossRef.
- S. Liang and H.-W. Su, Ind. Eng. Chem. Res., 2009, 48, 5558–5562 CrossRef.
- M. F. Desrosiers and J. M. Puhl, Radiat. Phys. Chem., 2009, 78, 461–463 CrossRef CAS.
- L. A. W. Abdou, O. A. Hakeim, M. S. Mahmoud and A. M. El-Naggar, Chem. Eng. J., 2011, 168, 752–758 CrossRef CAS.
- C. Jiang-Lin, L. Wen-Hua, Z. Jian-Qing and C. Chu-Nan, Acta Phys.-Chim. Sin., 2004, 20, 735–739 Search PubMed.
- Y. Guo, J. Zhou, X. Lou, R. Liu, D. Xiao, C. Fang, Z. Wang and J. Liu, Chem. Eng. J., 2014, 254, 538–544 CrossRef CAS.
- B. Kasprzyk-Hordern, M. Ziolek and J. Nawrocki, Appl. Catal., B, 2003, 46, 639–669 CrossRef CAS.
- A. R. Tehrani-Bagha, N. M. Mahmoodi and F. M. Menger, Desalination, 2010, 260, 34–38 CrossRef CAS.
- J. Hoigné and H. Bader, Water Res., 1976, 10, 377–386 CrossRef.
- A. Aleboyeh, H. Aleboyeh and Y. Moussa, Dyes Pigm., 2003, 57, 67–75 CrossRef CAS.
- P. Canizares, R. Paz, C. Saez and M. A. Radrigo, J. Environ. Manage., 2009, 90, 410–420 CrossRef CAS PubMed.
- J. C. Kruithof, IUVA News, 2005, 7, 17–20 Search PubMed.
-
(a) B. J. Martijn, P. C. Kamp and J. C. Kruithof, UV/H2O2 treatment an essential barrier in a multi barrier approach for organic contaminant control, PWN Publications, http://www.pwntechnologies.nl/resources/papers;
(b) B. J. Martijn, J. C. Kruithof and L. P. M. Rosenthal, Design and implementation of UV/H2O2 treatment in a full scale drinking water treatment plant, PWN Publications, http://www.pwntechnologies.nl/resources/papers.
- G. F. Ijpelaar, D. J. H. Harmsen and M. Heringa, Techneau Report, D2.4.1.1, May, 2007 Search PubMed.
- IAEA Scientific Report, Radiation Processing : Environmental Applications, 2007, ISBN 92-0-100507-5, IAEA, Vienna Search PubMed.
- IAEA Scientific Report, Radiation Treatment of Polluted Water and Wastewater, 2008, IAEA-TECHDOC-1598, IAEA, Vienna Search PubMed.
- W. J. Cooper, R. D. Curry and K. E. O'Shea, Environmental applications of ionizing radiation, John Wiley, 1998 Search PubMed.
- B. Han, J. Ko, J. Kim, Y. Kim, W. Chung, I. E. Makarov, A. V. Ponomarev and A. K. Pikaev, Radiat. Phys. Chem., 2002, 64, 53–59 CrossRef CAS.
- B. Han, J. K. Kim, Y. Kim, J. S. Choi and K. Y. Jeong, Radiat. Phys. Chem., 2012, 81, 1475–1478 CrossRef CAS.
- Y. A. Maruthi, N. L. Das, K. Hossain, K. S. S. Sarma, K. P. Rawat and S. Sabharwal, Afr. J. Environ. Sci. Technol., 2011, 5, 545–552 CAS.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 





