
CO2 adducts of Lewis acid–base pairs (LBCO2LA; LB = PMe3, NHC and LA = AlH3, AlCl3, BH3) − analogous to carboxylic acids and their derivatives†
Indu Purushothaman,
Susmita De* and
Pattiyil Parameswaran*
Department of Chemistry, National Institute of Technology Calicut, NIT Calicut Campus P.O., Kozhikode–673 601, Kerala, India. E-mail: param@nitc.ac.in; susmitade1980@gmail.com
First published on 5th November 2014
Abstract
The relationship between the structure and bonding of two different classes of molecules helps to understand and correlate their physiochemical activity. Here, we report the structure-bonding analogy between CO2 adducts of a Lewis acid (LA)–Lewis base (LB) pairs, LBCO2LA (LB = PMe3 and NHC; LA = AlH3, AlCl3 and BH3) and carboxylic acids and their derivatives, RCO2R′ (R, R′ = alkyl, H) by quantum mechanical calculations. The direction of charge flow in LBCO2LA is from LB to LA, whereas the reverse direction of charge flow (R′ to R) is observed for RCO2R′ leading to a formally negatively charged CO2 group (2A1) in both systems. This negatively charged bent CO2 group plays a deterministic role towards its bonding interaction with other fragments. The bonding analysis by the EDA-NOCV method indicates that both the LB and R groups form an electron sharing bond with the carbon atom of the bent CO2 fragment, whereas both LA and R′ form a donor–acceptor interaction with the oxygen atom. Our analysis suggests that the CO2 adducts of the Lewis acid (LA)–Lewis base (LB) pairs, LBCO2LA, can be considered as inorganic analogues of carboxylic acids and their derivatives, RCO2R′.
Introduction
The structure–activity relationship has a very prominent role in chemistry, which is based on the concept of understanding the physiochemical activity of a compound in relation to its molecular structure. The terms isoelectronic, isostructural, isolobal, isoreactive etc. are commonly used to correlate and draw a parallel between different types of molecules.1 The similarities between benzene and borazine,2 ethane and ammonia–borane adduct,2 the diagonal relationship between the first row and the second row elements viz., boron and silicon,3 carbon and phosphorous4 are some of the classical examples. Here, we report the structure-bonding analogy between CO2 adducts of Lewis acid (LA)–Lewis base (LB) pairs, LBCO2LA, as well as carboxylic acids and their derivatives, RCO2R′, (Scheme 1a). | ||
| Scheme 1 Schematic representation of the analogy between the experimentally reported (a) carboxylic acids, its derivatives and CO2 adducts of LA–LB pairs, (b) carboxylate anions and LB coordinated CO2 and (c) protonated carboxylic acids, bis-LA coordinated carboxylate anion and adduct of CO2 with two LAs and one LB. The ‘≡’ sign indicates structural equivalence. | ||
It has been reported that the sterically demanding frustrated LA–LB adducts show high reactivity towards a variety of small molecules such as inert CO2 at ambient conditions.5 Recently, CO2 activation by less sterically demanding LA–LB pairs (Me2PCH2AlMe2)2 and (Ph2PCH2AlMe2)2 by Fontaine and co-workers are also reported.6 These adducts show bent geometry for CO2 as that of the carboxylic acid derivatives RCO2R′ (Scheme 1a). In addition to the apparent structural similarities, reports of several adducts in the literature (Scheme 1b and c) indicate that LBCO2LA and RCO2R′ might possess similarity in the reactivity as well.7
Note that, the CO2 adduct of N-heterocyclic carbene, which can be considered as analogous to carboxylate anion (Scheme 1b)7a have been synthesized. In addition, CO2 adduct of two LAs and one LB, [R3PCO2(AlX 3)2]5b,c,h as well as the mixed adduct of the carboxylate anion with two LAs, RCO2[B(C6F5)3]2−,7b,c which are analogous to the protonated carboxylic acid (Scheme 1c) have also been reported. These examples show possible corroboration for the structure-bonding analogy between carboxylic acid and CO2 adduct of LA–LB pairs. However, the usual bonding description between the R, R′ group and CO2 in RCO2R′ is considered to be a covalent electron sharing. On the other hand, the LB and LA are commonly known to form donor–acceptor bonds and thus the CO2 adduct of the LA–LB pairs could also be considered as space separated donor–acceptor complex.8 Therefore, the following logical questions arise. Whether the bonding interaction of CO2 with the rest of the fragments in these two structurally similar classes of compounds is also similar? Could the CO2 adduct of LA–LB pairs be considered as a space separated donor–acceptor complex?
In order to explore the nature of bonding of CO2 fragment with LA and LB in LBCO2LA, we have carried out the quantum mechanical calculations at the BP86/TZ2P level of theory9,10 on the CO2 adduct of LA–LB pairs (LBCO2LA) (1–6, Fig. 1), where LB (LB = PMe3 and NHC) is coordinated to the C-atom and LA (LA = AlH3, AlCl3 and BH3) is coordinated to one of the O-atom of bent CO2. The bonding pattern of CO2 with LA and LB in LBCO2LA have been compared with the corresponding bonding pattern in carboxylic acids and derivatives, RCO2R′, where R and R′ = H and CH3 (7–9, Fig. 1). We have analysed the bonding situation in these two apparently different classes of molecules in detail by the EDA method,11 which has been successfully employed by Frenking and co-workers to understand the best bonding description.12
 | ||
| Fig. 1 Optimized geometries (BP86/TZ2P) and important geometrical parameters of Me3PCO2AlH3 (1), Me3PCO2AlCl3 (2), Me3PCO2BH3 (3), NHCCO2AlH3 (4), NHCCO2AlCl3 (5), NHCCO2BH3 (6), HCO2H (7), CH3CO2H (8) and CH3CO2CH3 (9). Distances are given in Å and angles are given in deg. Point group symmetry are given in parenthesis. | ||
Results and discussion
The equilibrium geometries of Me3PCO2AlH3 (1), Me3PCO2AlCl3 (2), Me3PCO2BH3 (3), NHCCO2AlH3 (4), NHCCO2AlCl3 (5), NHCCO2BH3 (6), HCO2H (7), CH3CO2H (8) and CH3CO2CH3 (9) at the BP86/TZ2P level of theory are shown in Fig. 1.9,10The geometrical parameters of 1–9 are reasonably close to the available experimentally reported values, which supports the adequacy of the level of theory used (Table S4†).5c,g,h,i The coordinated C–O bonds are slightly longer, whereas the uncoordinated C–O bonds are slightly shorter in RCO2R′ (7–9) as compared to LBCO2LA (1–6). Note that, the CO2 group in LBCO2LA is significantly bent similar to that in RCO2R′. The bending angle in 1–6 is in the range of 128.7–134.2°, which is slightly larger than those in RCO2R′, 7–9 (122.4–125.4°). Since the CO2 fragment is significantly bent in both the LBCO2LA adducts and RCO2R′, the bonding in these molecules can be best analysed by the interaction of the bent CO2 fragment with LB and LA in 1–6 as well as with R and R′ in 7–9.13 A correlation diagram connecting the important molecular orbitals (MO) of linear and bent geometries of singlet CO2 reveals the nature of the frontier orbitals, which can interact with LB and R as well as with LA and R′ (Fig. 2). The linear CO2 possesses a pair of mutually perpendicular degenerate non-bonding HOMO (−10.71 eV) having coefficient on the terminal O-atoms. It also has degenerate antibonding π* LUMO+1 (0.85 eV) with major coefficient on the central carbon atom.14 Each of the two mutually perpendicular set of π-MOs of bent CO2 can be considered as similar to the 3c–4e π-MOs in the allylic anion.
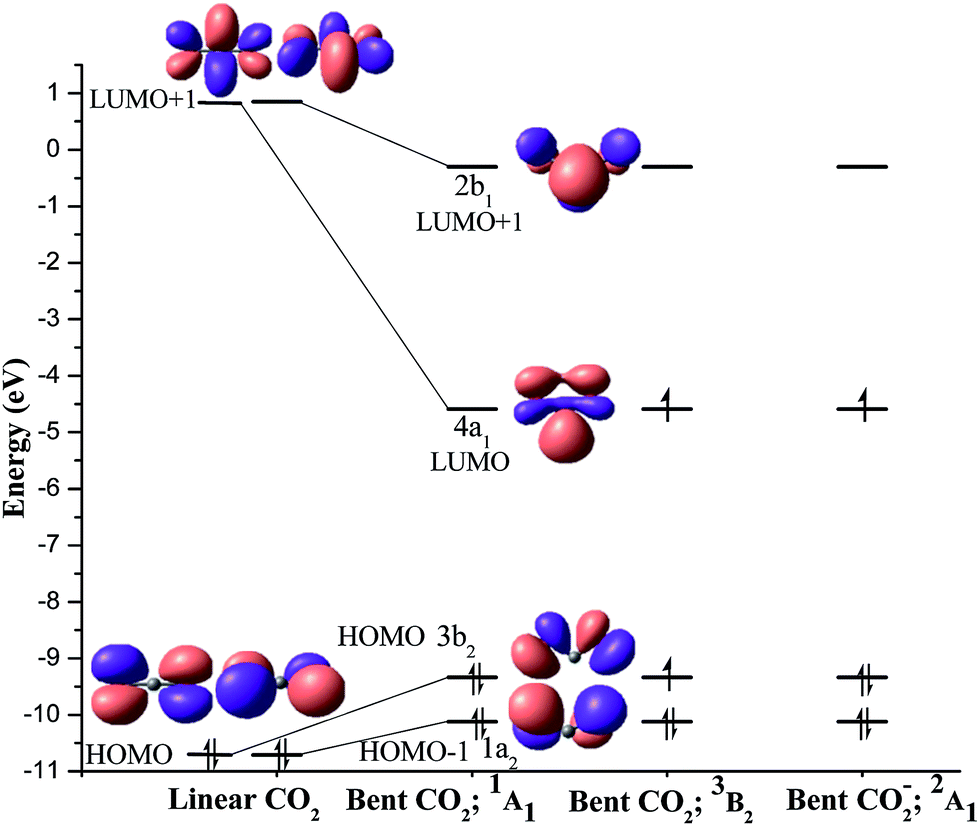 | ||
| Fig. 2 Correlation diagram connecting the frontier molecular orbitals of linear CO2 and singlet bent CO2 (1A1) at the M06/def2-TZVPP//BP86/def2-TZVPP level of theory.13 The nature of the molecular orbitals of the triplet bent CO2 (3B2) and doublet bent CO2− (2A1) are also shown. | ||
The HOMO of linear CO2 is destabilized in bent CO2 (3b2 and 1a2) due to the increased antibonding interaction between the p-orbitals on the O-atoms. This implies enhancement in the nucleophilic character of the terminal O-atoms. On the other hand, one of the degenerate π*-MOs in linear CO2 is highly stabilized (4a1, −4.59 eV) in the bent CO2 as compared to the other (2b1, −0.30 eV; Fig. 2). This stabilization is attributed to the enhanced bonding interaction in the in-plane MO (4a1, LUMO). Hence, the electrophilic character on the central carbon atom also enhances in bent CO2 as compared to linear CO2. Note that, the extent of stabilization of LUMO+1 is much higher than the destabilization of HOMO, which indicates significant enhancement of the electrophilic character in bent CO2 as compared to the nucleophilic character. The increased philicity of bent CO2 in (PPh3)2CCO2 has also been reported by Frenking and co-workers.15 Hence any factor, which causes the bending of CO2, increases the reactivity and would therefore favour the functionalization.
Apparently, the bonding description of the LBCO2LA adducts (1–6) can be assumed as the donation of the lone pair from LB to the electrophilic carbon atom of the bent CO2 (4a1, LUMO, Fig. 2) as well as the donation of the lone pair from oxygen atom of bent CO2 (3b2, HOMO, Fig. 2) to the empty orbital of LA. On the other hand, the bent CO2 group in RCO2R′ can be assumed in the singly excited state (3B2, Fig. 2) that can form electron sharing bonds with R and R′.16 In order to verify the best bonding description for the interaction of the bent CO2 with the rest of the fragments in 1–9, we have carried out detailed bonding analysis by NBO17 and EDA-NOCV methods.11,18
NBO group charge analysis in 1–6 (Table 1) shows high positive charge on LB and the negative charge is distributed over CO2 and LA, where CO2 carries higher negative charge (−0.44 to −0.72e) than LA (−0.14 to −0.33e). On the other hand, the R′ group in 7–9 (0.31 to 0.49e) carries higher positive charge than the R group (−0.01 to 0.11e). Hence, the net charge flow in 7–9 is from the R′ to R via CO2 group, where CO2 group acquires a negative charge (−0.30 to −0.60e). The direction of charge flow in LBCO2LA is from LB to LA, whereas the reverse direction of charge flow (R′ to R) is observed for RCO2R′ suggesting a formally negatively charged CO2 fragment (2A1, 1a223b224a11) in these molecules. This is consistent with the MO analysis that the bent CO2 has higher electrophilicity than nucleophilicity.15
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PMe3 | 0.85 | PMe3 | 0.88 | PMe3 | 0.79 | NHC | 0.76 | NHC | 0.72 | NHC | 0.75 | H | 0.11 | CH3 | −0.01 | CH3 | −0.01 |
| CO2 | −0.71 | CO2 | −0.72 | CO2 | −0.46 | CO2 | −0.61 | CO2 | −0.63 | CO2 | −0.44 | CO2 | −0.60 | CO2 | −0.48 | CO2 | −0.30 |
| AlH3 | −0.14 | AlCl3 | −0.16 | BH3 | −0.33 | AlH3 | −0.15 | AlCl3 | −0.09 | BH3 | −0.31 | H | 0.49 | H | 0.49 | CH3 | 0.31 |
If LBCO2LA (1–6) is a space separated donor–acceptor complex,8 the bonding interaction of CO2 with LB and LA fragments could be considered as donor–acceptor type viz., LB → CCO2LA and LBCO2O → LA. On the other hand, if the CO2 fragment in 7–9 is assumed to form electron sharing bonds with R and R′, the bonding interaction would then be an electron sharing type viz., R–CCO2R′ and RCO2O–R′.19 Here, we have explored the possibility of both electron sharing as well as donor–acceptor type bonding of CO2 group with other fragments in 1–9 (Scheme 2). The appropriate formal charges are assigned to the corresponding fragments to maintain the correct electronic configuration. The bond formed between the LB and C-atom of CO2LA can be of two types. The first possibility is the donor–acceptor interaction from the lone pair of LB to the empty orbital of the CO2LA fragment (similar to the LUMO, 4a1 of bent CO2, Fig. 2), which is represented as LB → CCO2LA in Scheme 2a and c.
 | ||
| Scheme 2 Schematic representation of the different types of bonding interactions of the CO2 fragment in LBCO2LA (1–6) and in RCO2R′ (7–9).20 | ||
The second possibility is the electron sharing ylidic-type interaction, which is represented as LB+–CCO2LA− in Scheme 2b and d. The electronic state of the fragments involved in these schemes are formed by transfer of one electron from the lone pair of LB to the empty orbital of CO2LA (similar to the electronic state 2A1 of bent CO2−, Fig. 2), which results the formally positively charged LB+ and the negatively charged CO2LA− fragments.16 In either way, once LB is bonded to the in-plane LUMO of bent CO2 (4a1), the in-plane π-MOs can reorganize as in-plane lone pair orbital on each O-atom. The O-atom can donate the lone pair to the empty orbital on LA, which is represented in Scheme 2a and b. There may be another possibility that one electron from the lone pair on the O-atom (similar to 3b2, Fig. 2) can be transferred to the vacant orbital on LA, which results LA− and LBCO2+ fragments having unpaired electrons. The interaction of LBCO2+ and LA− fragments results an ylidic type electron sharing LBCO2O+–LA− bond as shown in Scheme 2c and d. Similarly, two types of bonding interactions for the formation of bond between R and CO2R′ as well as RCO2 and R′ in 7–9 were also explored (Scheme 2e–h, Table S6†). The pertinent results of the EDA-NOCV analysis for the above mentioned bonding interactions (Scheme 2) are discussed next.
The complete EDA-NOCV results of 1, 4 and 9 are given in Table 2 and the EDA-NOCV results of the remaining molecules are similar and given in the ESI (Tables S1 and S2†). The electron sharing interactions for the ylidic type PMe3P+–CCO2AlH3− (ΔEorb = −153.5 kcal mol−1) in 1 and NHCC+–CCO2AlH3− (ΔEorb = −219.0 kcal mol−1) bonds in 4 (Scheme 2b and d) as well as electron sharing CH3C–CCO2CH3 bond (ΔEorb = −210.8 kcal mol−1) in 9 (Scheme 2f and h) has lower negative ΔEorb values as compared to the corresponding donor–acceptor interactions. The ΔEorb is the stabilization energy obtained by the overlap of the orbitals of the interacting fragments. The closer the electronic states of the interacting fragments to that in the molecule, the lesser will be the stabilization energy ΔEorb. Hence, the interacting fragments, which lead to the lowest ΔEorb can be considered as the best bonding fragments in terms of their electronic state.12
| Bond | 1 | 4 | 9 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P → C | P+–C− | O → Al | O+–Al− | C → C | C+–C− | O → Al | O+–Al− | C− → C+ | C–C | O− → C+ | O–C | |
| a Values in parenthesis give the percentage contribution to the total attractive interactions ΔEelstat + ΔEorb.b Values in parenthesis give the percentage contribution to the orbital interaction ΔEorb.c ΔErest = ΔEorb − (ΔEσ + ΔEπ). | ||||||||||||
| ΔEint | −66.4 | −162.7 | −39.8 | −218.7 | −85.8 | −196.1 | −36.4 | −233.9 | −332.3 | −105.7 | −280.6 | −99.2 |
| ΔEPauli | 243.4 | 197.3 | 69.3 | 209.6 | 397.1 | 267.4 | 56.4 | 181.3 | 404.9 | 265.6 | 189.8 | 359.4 |
| ΔEelstata | −136.8 (44.2%) | −206.6 (57.4%) | −65.8 (60.4%) | −199.3 (46.5%) | −222.5 (46.1%) | −244.5 (52.8%) | −57.9 (62.5%) | −179.0 (43.1%) | −380.5 (51.6%) | −160.5 (43.2%) | −248.9 (52.9%) | −159.8 (34.8%) |
| ΔEorba | −173.1 (55.8%) | −153.5 (42.6%) | −43.2 (39.6%) | −228.9 (53.5%) | −260.4 (53.9%) | −219.0 (47.2%) | −34.8 (37.5%) | −236.2 (56.9%) | −356.7 (48.4%) | −210.8 (56.7%) | −221.4 (47.1%) | −298.9 (65.2%) |
| ΔEσb | −144.6 (83.5%) | −123.9 (71.6%) | −22.7 (52.5%) | −211.5 (92.4%) | −213.4 (82.0%) | −182.3 (83.2%) | −21.2 (60.9%) | −225.9 (95.6%) | −314.0 (88.0%) | −184.8 (87.7%) | −174.8 (79.0%) | −279.9 (93.6%) |
| ΔEπb | −6.0 (3.5%) | −6.5 (3.8%) | −5.7 (13.9%) | −3.4 (1.5%) | −16.4 (6.3%) | −12.2 (5.6%) | −4.4 (12.6%) | −3.7 (1.6%) | −16.8 (4.7%) | −9.9 (4.7%) | −18.0 (8.1%) | −7.7 (2.6%) |
| ΔErestb,c | −22.5 (13.0%) | −42.7 (24.7%) | 14.8 (34.3%) | −14.0 (6.1%) | −30.6 (11.8%) | 35.5 (16.2%) | 9.2 (26.4%) | −6.6 (2.8%) | −25.9 (7.3%) | 16.1 (7.6%) | −28.6 (12.9%) | −11.3 (3.8%) |
| ΔEprep | 51.8 | 148.0 | 22.5 | 201.4 | 50.4 | 160.7 | 7.5 | 205.0 | 239.5 | 13.9 | 199.4 | 18.1 |
| ΔE(−De) | −14.7 | −14.7 | −17.3 | −17.3 | −35.4 | −35.4 | −28.9 | −28.9 | −92.8 | −92.8 | −81.1 | −81.1 |
| ΔEorb/ΔEelstat | 1.27 | 0.74 | 0.65 | 1.15 | 1.17 | 0.90 | 0.60 | 1.32 | 0.94 | 1.31 | 0.89 | 1.87 |
Hence, the bonding interaction of LB with CO2LA can be best represented as 1,2-dipolar electron sharing interaction, in which LB carries a formal positive charge and the C-atom carries a formal negative charge (Scheme 2b and d). This bonding representation is consistent with the high value for the preparation energy of the fragments (Table 2 and S1†) and the positive NBO charge (Table 1) for the LB fragments. The ΔEorb values also suggest that the classical electron sharing interaction between R and CO2R′ is indeed the best bonding representation (Scheme 2f and h). The comparable ΔEorb for the NHCC–CCO2AlH3 bond in 4 and CH3C–CCO2CH3 bond in 9 indicates that NHC can be considered as an appropriate substitution for the alkyl group for its coordination with CO2 molecule. This is supported by the experimental report of NHCCO2 (Scheme 1b), which is structurally equivalent to the carboxylate anion.7a The preference for the electron sharing interaction of LB and R group with CO2LA and CO2R′ is duly reflected in the percentage of electrostatic contribution (ΔEelstat). Note that, the LB–CCO2LA bonds in 1–6 has higher percentage of electrostatic contribution (51.2–57.8%) as compared to covalent contribution, whereas the reverse trend (ΔEelstat = 31.4–43.2%) is observed for the R–CCO2R′ bonds in the carboxylic acids and its derivatives, 7–9. The ratio ΔEorb/ΔEelstat (Table S1†) for compounds 1–6 is less than one (varies from 0.73 to 0.95) and that for compounds 7–9 is higher than one (varies from 1.31 to 2.19). The ratio ΔEorb/ΔEelstat have been used earlier by Frenking and co-workers for describing the dative bond in Fischer carbenes and covalent bond in Schrock carbenes.12a,b Although LB+–CCO2LA− and R–CCO2R′ bonds are best represented by electron sharing interactions, the nature of the bonds in 1–6 varied significantly in terms of ΔEorb/ΔEelstat as compared to that in 7–9.
The α-NOCV pair of orbitals (Ψ−1/Ψ1), deformation density plot (Δρ1) and the corresponding energy (ΔEσ) towards the dipolar electron sharing bond in 1 (ΔEσ = −123.9 kcal mol−1) and 4 (ΔEσ = −182.3 kcal mol−1) are shown in Fig. 3a and c. It clearly indicates the depletion of the electron density from LB+ and C-atom in CO2LA− as well as accumulation of electron density at the centre of the LB+−CCO2LA− bond. Similar plot of the deformation density in 9 (ΔEσ = −184.8 kcal mol−1) is obtained for the CH3C–CCO2CH3 bond as well (Fig. 3e). This is a typical deformation density plot for the electron sharing σ-bonds.18 The contribution from the π-type interaction is very negligible (Table 2, Fig. S1 and S2†). The ΔEπ comes from the hyperconjugative donation of the electrons from the perpendicular π-MO of the CO2LA−/CO2R′ fragment to the P–CMe σ*-MO of PMe3 in 1 (ΔEπ = −6.5 kcal mol−1) and C–H σ*-MO in 9 (ΔEπ = −9.9 kcal mol−1). However, the stronger π-acceptor NHC+ (vacant p-orbital) contributes to the higher ΔEπ (−12.2 kcal mol−1) in 4. It is noteworthy that, the interaction energy between the LB+ and CO2LA− is significantly higher in 1 and 4 than that in carboxylic acid derivatives, 9. However, the calculated bond dissociation energies (De) are significantly low in 1 and 4 as compared to 9. The low value of De in 1 and 4 is attributed to the higher preparation energy required for promoting the fragments in their respective electronic excited states.
 | ||
| Fig. 3 The α-NOCV pair of orbitals (Ψ−1/Ψ1) with their eigen values in parenthesis, the deformation density plots (Δρ1) and the corresponding orbital stabilization energies ΔEσ (kcal mol−1) for the (a) Me3PP+–C−CO2AlH3 bond and (b) Me3PCO2O → AlAlH3 bond in 1, (c) NHCC+–C−CO2AlH3 bond and (d) NHCCO2O → AlAlH3 bond in 4, (e) CH3C–CCO2CH3 bond and (f) CH3CO2O− → C+CH3 bond in 9 at the BP86/TZ2P level of theory. The direction of the charge flow is from red → blue. Isosurface value for NOCV pair of orbitals is 0.04 and that for the deformation density is 0.003. | ||
The ΔEorb for the donor–acceptor interaction LBCO2O → LA in 1 (ΔEorb = −43.2 kcal mol−1), 4 (ΔEorb = −34.8 kcal mol−1) and RCO2O− → R′+ in 9 (ΔEorb = −221.4 kcal mol−1) have lower negative values as compared to the corresponding electron sharing interactions (Table 2). Hence, the bonding interaction between O-atom in LBCO2 and LA can be considered as regular donor–acceptor interaction (Scheme 2a and b) and the bond formation between RCO2O and R′ in RCO2R′ can be best represented as 1,2-diploar donor–acceptor type interaction (Scheme 2e and f).21 Note that, according to the best bonding description of the carboxylic acids 7 and 8, the H-atom possesses a formal positive charge, which is consistent with the fact that the hydrogen atom of carboxylic acid is acidic. The EDA-NOCV data (Table 2 and S2†) suggest that the percentage of electrostatic contribution towards the donor–acceptor interaction for the RCO2O− → R′+ bonds in 7–9 is slightly higher than that for the LBCO2O → BBH3 bonds but lower than that for LBCO2O → AlAlH3/AlCl3 bonds in 1–6. The ΔEorb/ΔEelstat ratio (Table S2†) is in the range of 0.60 to 0.81 for LBCO2O → AlAlH3/AlCl3 bonds, 0.89 to 0.97 for RCO2O− → R′+ bonds and 1.09 and 1.11 for LBCO2O → BBH3 bonds. The variation of nature of the bonds in terms of ΔEorb/ΔEelstat ratio is less pronounced as compared to LB+–CCO2LA− and R–CCO2R′ bonds.12a,b
The α-NOCV pair of orbitals, deformation density plot (Δρ1) and stabilization energy (ΔEσ) for the charge transfer from the PMe3CO2 and NHCCO2 fragment to the AlH3 fragment in 1 and 4 are shown in Fig. 3b and d. Fig. 3f gives the plot of deformation density for the interaction of CH3CO2− with CH3+ fragment in 9. These plots clearly show depletion of electron density from the O-atom of the CO2 and the accumulation of the electron density at the Al-atom in 1 and 4 and C-atom in 9. These plots are similar to the typical plot for the donor–acceptor interactions.18 The ΔEσ for the RCO2O− → R′+ bond in 7–9 is the highest, followed by the LBCO2O → BBH3 bond in 3, 6 and the LBCO2O → AlAlH3/AlCl3 bond in 1, 2, 4 and 5 (Fig. 3 and S2†) indicating a stronger σ-interaction in the carboxylic acid and derivatives. Even though, the ΔEprep for the fragments involved for the RCO2O− → CR′+ bond formation in 7–9 is higher than those in 1–6, the calculated bond dissociation energies (De) for the RCO2O− → CR′+ bond is also higher in 7–9. This indicates that the acidic character (release of LA) of LBCO2LA is more than the acidic character (release of H+) of carboxylic acid. The experimental report of NHCCO2 (Scheme 1b)7a as well as RCO2(LA)2 (ref. 7b and c) and LBCO2(LA)2 (ref. 5b, c and h) (Scheme 1c) is noteworthy in this regard.
Hence, the EDA-NOCV data suggest that the best bonding description of LBCO2LA (1–6) can be represented by considering bond formation among LB+, CO2− and LA fragments as in Scheme 3a (Scheme 2b), whereas that of RCO2R′ (7–9) can be represented by considering bond formation among R, CO2− and R′+ fragments as in Scheme 3b (Scheme 2f). Note that, both EDA-NOCV and NBO charge (Table 1) analysis indicate that the bent CO2 group in LBCO2LA and RCO2R′ can be regarded as having a formal negative charge with the electronic state 2A1 (1a223b224a11; Fig. 2). This electronic state in the bent CO2− forces the groups attached to the C-atom to form electron sharing bond and the groups attached to the O-atom to form donor–acceptor bond. As a result, LB+ and R group form electron sharing LB+–CCO2LA− bond in LBCO2LA and R–CCO2LA bond in RCO2R′, whereas the LA and R′+ group form donor–acceptor LBCO2O → LA bond in LBCO2LA and LBCO2O− → R′+ bond in RCO2R′. Hence, the bent geometry and the electronic state of CO2 plays a deterministic role towards the bonding interaction with the other groups by altering the usual bonding pattern of LB and R′. Therefore, the CO2 adduct of LA–LB pair LBCO2LA can not be classified as space-separated donor–acceptor complexes, rather it can be considered as inorganic analogue of carboxylic acid and its derivatives, RCO2R′.
 | ||
| Scheme 3 Schematic representation of the bonding interactions of the bent CO2 fragment with other fragments in (a) LBCO2LA (1–6) and (b) RCO2R′ (7–9). | ||
Conclusions
The structure and bonding of the CO2 adduct of Lewis acid (LA)–Lewis base (LB) pair, LBCO2LA (LB = PMe3 and NHC; LA = AlH3, AlCl3 and BH3) and carboxylic acid and its derivatives, RCO2R′ (R, R′ = alkyl, H) have been analyzed by quantum mechanical calculations at the BP86/TZ2P level of theory. The CO2 group in LBCO2LA is significantly bent, which is similar to that of carboxylic acid and derivatives. There is significant enhancement of the electrophilicity and nucleophilicity of bent CO2 as compared to linear CO2. However, the enhancement of electrophilicity is higher than the nucleophilicity. The NBO analysis indicates that the direction of charge flow in LBCO2LA is from LB to LA, whereas the reverse direction of charge flow, R′ to R, is observed for RCO2R′. The EDA-NOCV data suggest that the bonding interaction of LB with the C-atom of CO2LA in LBCO2LA can be best described as 1,2-dipolar LB+–CCO2LA− electron sharing bond. While, the bonding interaction of R with the C-atom of CO2R′ in RCO2R′ can be represented as classical electron sharing bond. On the other hand, the bonding interaction of the O-atom of LBCO2 with LA in LBCO2LA is a classical donor–acceptor bond. However, the bonding of the O-atom of RCO2 with R′ in RCO2R′ can be best represented as 1,2-dipolar RCO2O− → R′+ donor–acceptor interaction. The geometry and the electronic state of bent CO2 plays a deterministic role towards its bonding with the other groups in LBCO2LA and RCO2R′. Therefore, the CO2 adduct of LA–LB pair, LBCO2LA, can be considered as inorganic analogue of carboxylic acid and its derivatives, RCO2R′, although the nature of each type of bonds varied in terms of ratio of orbital to electrostatic contribution (ΔEorb/ΔEelstat).Computational methods
All the geometries were optimized at the gradient-corrected BP869 density functional with the basis set TZ2P.10 The calculations were performed using ADF 2013.01 package.22 Meta-GGA exchange–correlation functional M0623 with def2-TZVPP24 basis set was used for the single point calculation on geometries optimized at the BP86/def2-TZVPP level of theory using Gaussian 09 package.25 Natural bond order (NBO)17 calculations were computed at the same level of theory.The nature of LB–C bond, O–LA bond, R–C bond and O–R′ bond were investigated by EDA-NOCV analysis at the BP86/TZ2P level of theory using ADF 2013.01 program.22 Scalar relativistic effects were incorporated using Zeroth Order Regular Approximation (ZORA)26 and the core electrons were treated by the frozen-core approximations. Energy Decomposition Analysis (EDA)11 gives the instantaneous interaction energy (ΔEint) between two fragments in the frozen geometry of the compound. The interaction energy can be divided into three parts:
| ΔEint = ΔEelstat + ΔEPauli + ΔEorb |
ΔEelstat gives the electrostatic interaction energy between the frozen charge densities of the two fragments. ΔEPauli gives the repulsive interaction between two fragments, which are caused by the electrons of same spin. ΔEorb is the lowering in energy due to the overlap of orbitals of the two fragments. Sum of ΔEint and ΔEprep (energy necessary to promote the fragments from their ground state to the geometry and electronic state in the compound) gives −De (dissociation energy).
| −De = ΔEint + ΔEprep |
EDA-NOCV analysis is an extension of EDA analysis in which ΔEorb term is decomposed into the contributions from different natural orbitals for chemical valence (NOCV).18 The EDA-NOCV scheme gives insight to the orbital interactions by providing the energy contributions for each specific orbital interaction between fragments to the total bond energy. The deformation density, Δρ, which is incorporated with bond formation is partitioned into the different components (σ, π and δ) of the chemical bond.
NOCV scheme has been derived from Nalewajski–Mrozek valence theory as eigenvectors that diagonalize the deformation density matrix. The NOCV pairs (Ψ−k, Ψk) decompose the differential density Δρ into NOCV contributions (Δρk).
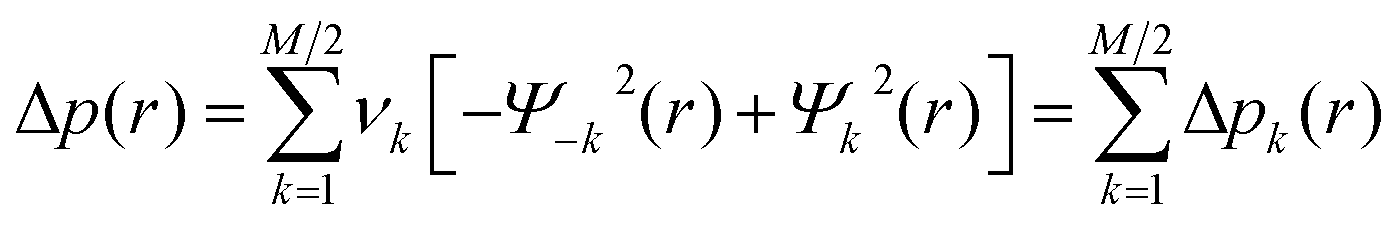
where FTSi,i are diagonal Kohn–Sham matrix elements defined over NOCV with respect to the transition state (TS) density (at the midpoint between density of the molecule and the sum of fragment densities). The ΔEorbk provide energetic estimation of Δρk that may be related to the importance of a particular electron flow channel for the bonding between considered molecular fragments.
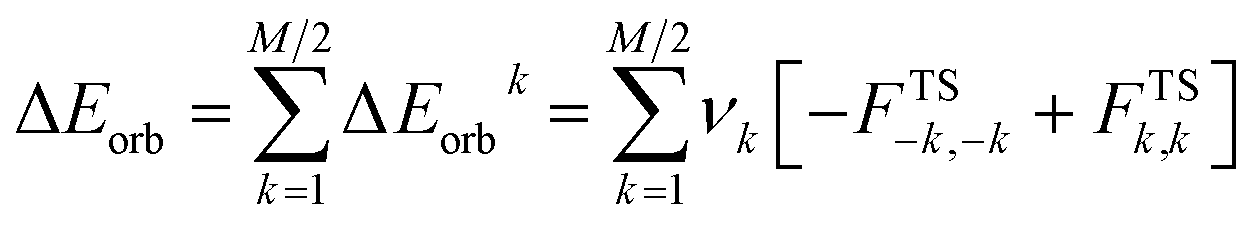
Acknowledgements
The authors acknowledge the financial support received from the Department of Science and Technology and INSA-INSPIRE. SD thanks for research grant, no. SR/FT/CS-42/2011 and IFA12-CH-76 for research grant and fellowship. PP thanks for research grant, no. SR/FT/CS-121/2011. IP thanks MHRD for Junior Research Fellowship.Notes and references
- (a) A. D. McNaught and A. Wilkinson, Compendium of Chemical Terminology, the “Gold Book”, Blackwell Scientific Publications, IUPAC, Oxford, 2nd edn, 1997 Search PubMed; (b) R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1982, 21, 711 CrossRef; (c) T. A. Albright, J. K. Burdett and M.-H. Whangbo, Orbital Interactions in Chemistry, John Wiley & Sons, Inc., New York, 2nd edn, 2013 Search PubMed.
- (a) J. E. Huheey, E. A. Keiter, R. L. Keiter and O. K. Medhi Inorganic chemistry: principles of structure and reactivity, Pearson Education, India, 2006 Search PubMed; (b) A. S. Phukan, R. P. Kalagi, S. R. Gadre and E. D. Jemmis, Inorg. Chem., 2004, 43, 5824 CrossRef CAS PubMed.
- (a) E. D. Jemmis, B. V. Prasad, S. Tsuzuki and K. Tanabe, J. Phys. Chem., 1990, 94, 5530 CrossRef CAS; (b) D. Mallick, P. Parameswaran and E. D. Jemmis, J. Phys. Chem. A, 2008, 112, 13080 CrossRef CAS PubMed; (c) E. D. Jemmis and P. Parameswaran, Chem.–Eur. J., 2007, 13, 2622 CrossRef CAS PubMed; (d) G. Subramanian, E. D. Jemmis and B. V. Prasad, Chem. Phys. Lett., 1994, 217, 296 CrossRef CAS; (e) G. Subramanian and E. D. Jemmis, Chem. Phys. Lett., 1992, 200, 567 CrossRef CAS.
- (a) K. B. Dillon, F. Mathey and J. F. Nixon Phosphorus: The Carbon Copy, Wiley, Chichester, 1998 Search PubMed; (b) F. Mathey, Angew. Chem., Int. Ed., 2003, 42, 1578 CrossRef CAS PubMed.
- (a) C. M. Mömming, E. Otten, G. Kehr, R. Fröhlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643 CrossRef PubMed; (b) G. Ménard and D. W. Stephan, J. Am. Chem. Soc., 2010, 132, 1796 CrossRef PubMed; (c) G. Ménard and D. W. Stephan, Angew. Chem., Int. Ed., 2011, 50, 8396 CrossRef PubMed; (d) X. Zhao and D. W. Stephan, Chem. Commun., 2011, 47, 1833 RSC; (e) M. J. Sgro, J. Dömer and D. W. Stephan, Chem. Commun., 2012, 48, 7253 RSC; (f) C. Appelt, H. Westenberg, F. Bertini, A. W. Ehlers, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2011, 50, 3925 CrossRef CAS PubMed; (g) R. C. Neu, G. Ménard and D. W. Stephan, Dalton Trans., 2012, 41, 9016 RSC; (h) G. Ménard, T. M. Gilbert, J. A. Hatnean, A. Kraft, I. Krossing and D. W. Stephan, Organometallics, 2013, 32, 4416 CrossRef; (i) E. Theuergarten, T. Bannenberg, M. D. Walter, D. Holschumacher, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2014, 43, 1651 RSC.
- J. Boudreau, M.-A. Courtemanche and F.-G. Fontaine, Chem. Commun., 2011, 47, 11131 RSC.
- (a) A. Tudose, A. Demonceau and L. Delaude, J. Organomet. Chem., 2006, 691, 5356 CrossRef CAS; (b) B.-H. Xu, R. A. A. Yanez, H. Nakatsuka, M. Kitamura, R. Frçhlich, G. Kehr and G. Erker, Chem.–Asian J., 2012, 7, 1347 CrossRef CAS PubMed; (c) A. E. Ashley, A. L. Thompson and D. O'Hare, Angew. Chem., Int. Ed., 2009, 48, 9839 CrossRef CAS PubMed; (d) A. Berkefeld, W. E. Piers and M. Parvez, J. Am. Chem. Soc., 2010, 132, 10660 CrossRef CAS PubMed.
- C. Goedecke, R. Sitt and G. Frenking, Inorg. Chem., 2012, 51, 11259 CrossRef CAS PubMed.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed; (b) J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef; (c) J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 7406 CrossRef.
- J. G. Snijders and E. J. Baerends, At. Data Nucl. Data Tables, 1981, 26, 483 CrossRef CAS.
- (a) K. Morokuma, J. Chem. Phys., 1971, 55, 1236 CrossRef CAS; (b) T. Zeigler and A. Rauk, Inorg. Chem., 1979, 18, 1755 CrossRef; (c) T. Zeigler and A. Rauk, Inorg. Chem., 1979, 18, 1558 CrossRef; (d) M. v. Hopffgarten and G. Frenking, WIREs Comput. Mol. Sci., 2012, 2, 43 CrossRef.
- (a) G. Frenking, K. Wichmann, N. Fröhlich, C. Loschen, M. Lein, J. Frunzke and V. M. Rayón, Coord. Chem. Rev., 2003, 238–239, 55 CrossRef CAS; (b) G. Frenking, M. Solà and S. F. Vyboishchikov, J. Organomet. Chem., 2005, 690, 6178 CrossRef CAS; (c) A. Krapp, K. K. Pandey and G. Frenking, J. Am. Chem. Soc., 2007, 129, 7596 CrossRef CAS PubMed; (d) A. Krapp, F. M. Bickelhaupt and G. Frenking, Chem.–Eur. J., 2006, 12, 9196 CrossRef CAS PubMed; (e) A. Krapp and G. Frenking, J. Am. Chem. Soc., 2008, 130, 16646 CrossRef CAS PubMed; (f) P. Parameswaran and G. Frenking, Chem.–Eur. J., 2009, 15, 8807 CrossRef CAS PubMed; (g) P. Parameswaran and G. Frenking, Chem.–Eur. J., 2009, 15, 8817 CrossRef CAS PubMed; (h) G. Frenking, R. Tonner, S. Klein, N. Takagi, T. Shimizu, A. Krapp, K. K. Pandey and P. Parameswaran, Chem. Soc. Rev., 2014, 43, 5106 RSC; (i) G. Frenking and F. M. Bickelhaupt in The Chemical Bond: Fundamental Aspects of Chemical Bonding, ed. G. Frenking and S. Shaik, Wiley-VCH Verlag GmbH & Co, Weinheim, Germany, 2014, ch. 4, p. 121 Search PubMed.
- The energy required for the geometrical distortion from linear geometry to singlet bent geometry of CO2 is 74.0 kcal mol−1 and to triplet bent geometry is 109.4 kcal mol−1 at the M06/def2-TZVPP//BP86/def2-TZVPP level of theory using Gaussian 09 program package. The bent geometry of CO2 is not a stationary point in the potential energy surface at BP86/TZ2P using ADF2013.01 program package. Hence, we have used the MO energies at the M06/def2-TZVPP//BP86/def2-TZVPP level of theory in Fig. 2.
- LUMOs are the degenerate antibonding π*-MOs at the M06/def2-SVP level of theory. It is same as the LUMO+1 at the M06/def2-TZVPP//BP86/def2-TZVPP level of theory.
- W. Petz, C. Kutschera, M. Heitbaum, G. Frenking, R. Tonner and B. Neumüller, Inorg. Chem., 2005, 44, 1263 CrossRef CAS PubMed.
- We have only considered one electron excitation for the fragments.
- (a) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS; (b) E. D. Glendening, A. E. Reed, J. E. Carpenter and F. WeinholdNBO Version 5.9 Search PubMed.
- (a) M. Mitoraj and A. Michalak, Organometallics, 2007, 26, 6576 CrossRef CAS; (b) M. Mitoraj and A. Michalak, J. Mol. Model., 2007, 13, 347 CrossRef CAS PubMed; (c) A. Michalak, M. Mitoraj and T. Zeigler, J. Phys. Chem. A, 2008, 112, 1933 CrossRef CAS PubMed; (d) M. Mitoraj and A. Michalak, J. Mol. Model., 2008, 14, 681 CrossRef CAS PubMed; (e) M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962 CrossRef CAS; (f) T. A. N. Nguyen and G. Frenking, Chem.–Eur. J., 2012, 18, 12733 CrossRef CAS PubMed; (g) M. Mousavi and G. Frenking, Organometallics, 2013, 32, 1743 CrossRef CAS.
- The conventional use of “→” bond in a donor–acceptor complex (LBLA) indicates donation of a lone pair of electron from LB to the empty orbital of LA (LB → LA). However, if the complex LBLA is described by the zwitterionic form it would yield a formal positive charge on LB and a formal negative charge on LA. The appropriate Lewis structure for LB and LA would then contain singly occupied orbitals, which forms the electron sharing bond (LB+–LA−). The use of “→” bond and “–” bond have been a point of discussion by Frenking and Krossing. See references: (a) D. Himmel, I. Krossing and A. Schnepf, Angew. Chem., Int. Ed., 2014, 53, 370 CrossRef CAS PubMed; (b) G. Frenking, Angew. Chem., Int. Ed., 2014, 53, 6040 CrossRef CAS PubMed.
- The dotted line indicates four π-electrons in the perpendicular plane similar to the bent CO2 and allylic anion.
- To confirm the nature of the alcoholic O–R′ bond in ester, 9, we have further carried out EDA-NOCV analysis on CH3CO2C2H5 (10) and C2H5CO2C3H7 (11) as well (Table S5†). The results are consistent with the conclusion drawn for CH3CO2CH3 (9). Hence, the alcoholic O–R′ bonds in the esters 9–11 can be considered as charge separated donor–acceptor RCO2O− → R′+. The comparable values for the ratio ΔEorb/ΔEelstat in 9 (0.89), 10 (0.91) and 11 (0.95) indicates similar bonding nature. Hence, the results of compound 9 are only presented in the text.
- ADF 2013.01, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, http://www.scm.com CrossRef CAS; G. teVelde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision B.01), Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- (a) Ch. Chang, M. Pelissier and Ph. Durand, Phys. Scr., 1986, 34, 394 CrossRef CAS; (b) J.-L. Heully, I. Lindgren, E. Lindroth, S. Lundquist and A.-M. Martensson Pendrill, J. Phys. B: At., Mol. Opt. Phys., 1986, 19, 2799 CrossRef CAS; (c) E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597 CrossRef CAS; (d) E. van Lenthe, J. G. Snijders and E. J. Baerends, J. Chem. Phys., 1996, 105, 6505 CrossRef CAS; (e) E. van Lenthe, R. van Leeuwen, E. J. Baerends and J. G. Snijders, Int. J. Quantum Chem., 1996, 57, 281 CrossRef CAS; (f) E. van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: It contains the optimized Cartesian coordinates, total bonding energies and EDA-NOCV data for all the molecules. See DOI: 10.1039/c4ra10269j |
| This journal is © The Royal Society of Chemistry 2014 |