DOI:
10.1039/C4RA06813K
(Paper)
RSC Adv., 2014,
4, 60413-60420
Synthesis of thermo-responsive polymer–protein conjugates through disulfide bonding†
Received
8th July 2014
, Accepted 6th November 2014
First published on 6th November 2014
Abstract
A novel alternative was developed for the synthesis of polymer–protein conjugates with a controllable number of polymer chains. Human serum albumin (HSA), as a model protein, was grafted by poly(N-isopropyl-acrylamide) (PNIPAM) using N-succinimidyl-3-(2-pyridyldithiol) propionate as a coupling agent at room temperature in aqueous media. The influence of grafting number on the stability of the protein–polymer conjugate against the degradation of enzyme was examined, and the temperature dependent bioactivity was monitored through testing the warfarin-binding affinity. Furthermore, the protein could be recovered by incubating the conjugate under reducing conditions to cause the cleavage of the multiple disulfide bonds.
Introduction
Stimuli-responsive polymer–protein conjugates, also called smart polymer–protein conjugates, have been extensively studied for their behavior to external stimuli such as redox changes, ultrasonication, and shifts in temperature, magnetic field, pH, and light. They can be used for drug/gene delivery, as biosensors, or imaging of therapeutic and diagnostic agents.1–7 Indeed, many stimuli-responsive polymer–protein conjugates have been used in clinical applications.8,9 In the early 1980s, Hoffman demonstrated that protein–PNIPAM conjugates block substrate binding to protein active sites; enzyme activity can be triggered by decreasing temperature below the lower critical solution temperature (LCST),10–13 e.g. β-D-glycosidase–PNIPAM is a thermo-reversible and phase-separating polymer–enzyme conjugate generated by reaction of the N-hydroxysuccinimide (NHS) ester functional end group to the amino residue of the protein.14 Similarly, streptavidin–PDEAAm (poly(N,N-diethylacrylamide)) conjugates exhibit temperature-dependent biotin binding; temperature modulation causes collapse of the polymer chain around the streptavidin active site.15 This method has been widely exploited to generate environment-responsive polymer–protein switches.16–21 For solubility purposes, some proteins were also coupled to PNIPAM block co-polymer chains.22–24 Polymers can easily be coupled to many bio-reactive groups, making it possible to direct attachment to proteins.25–35 Most polymer–protein conjugates are linked via the amino group as there are plenty of amino groups on the surface of a protein.36–46 Free cysteine groups are generally present on the protein surface at defined locations, and are often used to generate site-specific target polymer–protein conjugates;47–49 in addition, polymer–protein conjugates via cysteine groups containing disulfide bonds that can be reversibly reduced.50 Reduction-responsive nanoparticles have been widely used for intracellular drug delivery because of the relatively high concentration of intracellular reducing agents (glutathione).51–58 For instance, bovine serum albumin (BSA) contains only one free thiol group on cysteine no. 34; this residue was targeted for PNIPAM conjugation to make it temperature-sensitive.59 To obtain more free thiol groups for the reaction with the polymer, BSA can be reduced by tris(2-carboxyethyl)phosphine (TCEP) but retains its native properties.60 Lysosome amino groups have been used to obtain indirect disulfide bonds via thiazolidine-2-thione terminated polyPEGMA, conjugates of which were biodegradablility and activity could be triggered by incubation with TCEP.50 Most cysteine residues form intramolecular disulfide bonds to stabilize the native protein structure and properties. Thus, free cysteine is extremely rare in most native proteins. Proteins that do not naturally contain free cysteine have been genetically engineered to introduce cysteine into the protein sequence.61–65
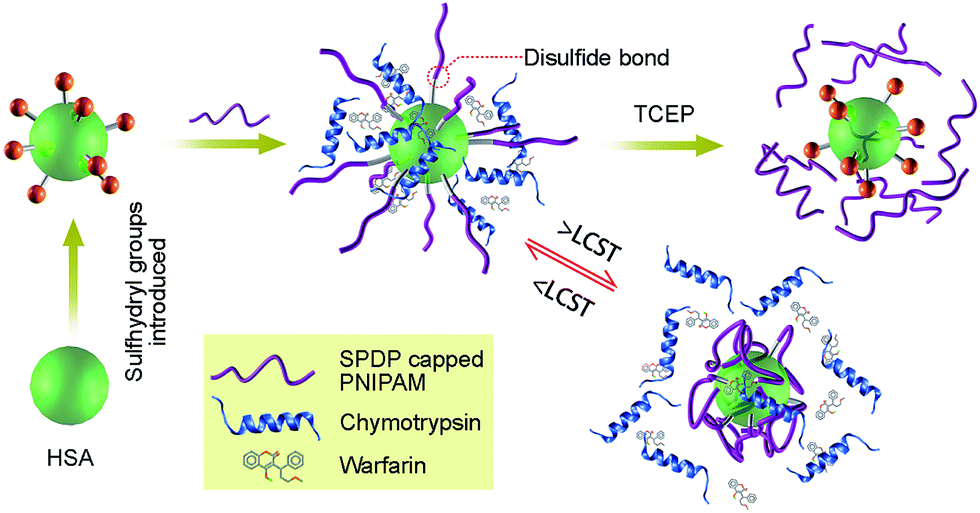
The molecular weight of PNIPAM is a key factor in controlling the conjugates behavior in addition to the number of grafting as discussed in the literatures.14,15,66 Nevertheless, in this work, we are focusing from another view which is to examine the influence of grafting density and conformation change of the polymer chains on the bioactivity of a protein. Herein, we report an alternative in which multiple disulfide bonds are used to connect PNIPAMs to human serum albumin (HSA), i.e. N-succinimidyl-3-(2-pyridyldithiol) propionate (SPDP) connects the amino group of HSA and amino-terminated PNIPAM to form disulfide bond between them. SPDP is a hetero-bifunctional cross-linking reagent with NHS (activated ester) and 2-pyridyldithio group that react with amine (NH2) and sulfhydryl (SH) group, respectively. Thus, intermolecular cross-bonds can easily be introduced between the protein and polymer without the concomitant formation of intramolecular cross-bonds.67 We produced thermo-responsive HSA–PNIPAM conjugates via indirect multi-disulfide bonds, which are stable under physiological condition and labile under mild reducing condition, and are thus promising for use in intracellular protein delivery. This novel method supplies an alternative to traditional methods of relying on proteins with few or no cysteine residues; we can produce reducible polymer-conjugates that may improve the development of polymer–protein systems (Scheme 1). Warfarin binding affinity for the conjugate was favored below the LCST of PNIPAM but was inhibited at temperatures above the LCST due to collapse of the polymer chains around the HSA, which efficiently prevented the approaching of warfarin. In comparison to the native HSA, the stability of the protein conjugates against proteolytic degradation was greatly enhanced. Warfarin binding affinity and stability against proteolytic degradation can be controlled by adjusting the number of coupled polymers. The polymer–protein conjugates own dual responsive, i.e. thermo- and reducing- responsive, which is a promising tool for sub-hypothermic therapy and intracellular protein delivery.
 |
| | Scheme 1 HSA–PNIPAM conjugates synthesized via multi-disulfide bonds exhibited thermo-responsive collapse to prevent warfarin/enzyme from approaching, thus prohibiting the protein reaction. This strategy can also be used to tailor protein properties by changing the amount of polymer during conjugate synthesis. Inclusion of the disulfide bonds allows dissociation under mild reducing conditions, making it a promising tool for intracellular protein delivery. | |
Experimental
Materials
Human serum albumin was purchased from Utech (USA). N-succinimidyl-3-(2-pyridyldithio) propionate (SPDP) was purchased from Thermo-Fisher Scientific. Chymotrypsin (MW-25 kDa) was purchased from VWR International (UK). Tris(2-carboxyethyl) phosphine hydrochloride (TCEP) was purchased from Strem Chemicals. Poly(N-isopropyl-acrylamide), amine terminated (PNIPAM–NH2), average Mn 5500, warfarin (3-(α-acetonylbenzyl)-4-hydroxycoumarin), dimethylformamide (DMF), phosphate-buffered saline (PBS), solvents, and other compounds were obtained from Sigma-Aldrich, USA.
Synthesis of multi-sulfhydryl HSA
HSA (100 mg, 1.51 μmol) was dissolved in 20 mL PBS (0.01 M, pH 7.4). To this was added 1 mL of SPDP (7.55 or 15.1 μmol) in DMF dropwise. The mixture was stirred for 2 h at room temperature. Then, TCEP (22.65 or 45.3 μmol) in 1 mL PBS (0.01 M, pH 7.4) was added dropwise and stirred for 2 h to obtain a multiple sulfhydryl-modified HSA solution. The solution was dialyzed using a membrane with a molecular weight cut-off of 14 kDa against Milli-Q water with six exchanges at room temperature for 48 h. A portion of the solution (solution 1) was retained for the following synthesis and the remainder was freeze-dried for characterization.
Synthesis of SPDP capped PNIPAM–NH2 (PNIPAM–SPDP)
PNIPAM–NH2 (average Mn 5500) (100 mg, 18.18 μmol) was dissolved in 10 mL PBS (0.01 M, pH 7.4). To this was added 1 mL of SPDP (9.09 μmol) in DMF. The mixture was stirred for 2 h at room temperature to obtain SPDP capped PNIPAM (solution 2).
Synthesis of multi-disulfide HSA–PNIPAM conjugates
Solution 2 (containing 25 or 50 equiv. PNIPAM–SPDP) was added to solution 1 (containing 1 equiv. protein) dropwise and stirred for 24 h at room temperature to obtain the crude PNIPAM–HSA conjugated product, which was transferred to a dialysis tube and dialyzed against Milli-Q water using a membrane with a molecular weight cut-off of 14 kDa, at room temperature for 48 h with six exchanges. The solution was centrifuged to remove any precipitate and then freeze-dried and re-dissolved in Milli-Q water and purified on a Sephadex G-75 gel permeation column (column length 0.3 m, internal diameter 2.2 cm). The protein-containing eluate was collected and freeze-dried to obtain a white powder-like product with yields of 70–76 wt%, calculated from the mass of the product to the protein feed. The products were stored at −20 °C until further characterization.
The chemical composition of the conjugates was determined by elemental analysis. The average number of grafted PNIPAM chains per HSA molecule was calculated as nPNIPAM = (MWconjugate − MWprotein)/MWPNIPAM, where MWconjugate, MWprotein, and MWPNIPAM are the weight mean molecular weights of the HSA–PNIPAM conjugates, HSA, and PNIPAM–NH2, respectively.
Characterization
Elemental analysis was performed on a Flash 2000-Thermo Scientific CHNO Analyzer. 1H NMR was recorded on a Bruker AV-III 600 MHz SB Liquid NMR Spectrometer (Bruker Corporation, Switzerland) by dissolving the samples in deuterated water (D2O). The size of the HSA–PNIPAM conjugates were measured on a Zetasizer (Nano Series, Malvern Instruments, UK) in aqueous solution at a fixed concentration of 0.2 mg mL−1. Circular dichroism (CD) was measured with a Jasco-815 spectropolarimeter (Jasco, Tokyo, Japan).
Biodegradation of HSA–PNIPAM conjugates by proteinase
Mini-PROTEAN TGX Precast Gel (4–20%), 10× Tris/glycine/SDS electrophoresis migration buffer, Precision Plus Protein Unstained Standards (10–250 kDa), and Bio-Safe colloidal Coomassie Brilliant Blue G-250 protein staining solution were purchased from Bio-Rad (USA). The HSA–PNIPAM conjugates were evaluated by incubating the conjugates with chymotrypsin in vitro. 1 mL HSA–PNIPAM conjugate solution or HSA containing 0.5 mg HSA equiv. was placed in 2 mL vials, mixed with 119 IU (0.119 mg) chymotrypsin, and held at 25 °C and 37 °C for 90 min. The solution was heated at 100 °C for 5 min to stop the reaction and then 0.5 mL TCEP (0.129 mg mL−1) was added to cleave the disulfide bond and release the HSA. One part sample was diluted with one part Laemmli sample buffer. SDS-PAGE was performed in a 4–20% precast polyacrylamide gel in a Mini-PROTEAN Tetra Cell (Bio-Rad, USA). Protein migration was achieved at a constant voltage of 100 V for about 1.5 h in migration buffer. After electrophoresis, the gel was stained with colloidal Coomassie Blue G250 with gentle agitation and then washed in Milli-Q water until the background was clear.
Warfarin binding by HSA–PNIPAM conjugates
The experiments were carried out as previously reported.68 The warfarin fluorophores were excited at 330 nm, and the fluorescence was measured at the emission maximum (approx. 380 nm). Typical assays were performed as follows: a preheated HSA or HSA–PNIPAM conjugate solution (100 μL, HSA concentration: 0.105 mM) in PBS (0.01 M, pH 7.4) and a preheated solution of warfarin (150 μL, 0.091 mM) were immediately pipetted into 96-well plates and vortexed for 60 s. The amount of HSA is equal in each well, and the solutions were incubated at the desired temperature for 15 min. To measure binding, the solution was excited at 330 nm and fluorescence was measured at 380 nm using a microplate reader (SpectraMax M5, Molecular Devices). Percent binding of the HSA–PNIPAM conjugate is reported relative to native HSA tested at the assay temperature. Fluorescence intensity was corrected for baseline (buffer + warfarin) fluorescence. All experiments were performed in triplicate; values are presented as means ± standard deviation.
The effect of thermal cycling on the warfarin binding affinity of HSA–PNIPAM conjugates
To investigate the effect of thermal cycling on the warfarin binding affinity of HSA–PNIPAM conjugates, assays were performed after multiple heating/cooling cycles. The temperature was raised from 25 °C to 37 °C (below and above the conjugate's LCST) and maintained at 37 °C for 30 min, and the binding affinity was measured after 10 min at 25 °C. The procedure was similar to that described above with the following exceptions. Seven samples of HSA–PNIPAM conjugates were prepared; the binding affinity of one sample was measured at 25 °C, as described above. The remaining samples were heated and cooled multiple times with one sample removed for measurement at 25 °C in each cycle. The process was repeated for a total of seven heating/cooling cycles. The warfarin binding affinity of HSA before recycling is defined as 100%.
Results and discussion
Synthesis of multi-sulfhydryl HSA and SPDP capped PNIPAM
Multi-sulfhydryl HSA was synthesized by reacting SPDP with free amino residues of HSA (Scheme 2). This reaction occurs readily and is specific for amino groups under mild conditions. The feed ratio of lysine residues in SPDP is ca. 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 12:1 due to the overall 59 lysine residues per HSA molecule. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectroscopy provided direct evidence of efficient protein functionalization with the SPDP (Fig. S1, ESI†). 2-Pyridyldisulfide-terminated PNIPAM, i.e. SPDP capped PNIPAM, was prepared by an esterification reaction between amino-terminated PNIPAM and SPDP in PBS (0.01 M, pH 7.4). The structure of SPDP capped PNIPAM, which was synthesized by changing the PNIPAM–NH2–SPDP feed ratio to 1:2, dialyzed, and then freeze-dried, was confirmed by 1H NMR (Fig. S2, ESI†). The appearance of pyridine protons, i.e. 8.4 ppm, 7.8 ppm and 7.3 ppm, indicates the formation of PNIPAM–SPDP.
:1 to 12:1 due to the overall 59 lysine residues per HSA molecule. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectroscopy provided direct evidence of efficient protein functionalization with the SPDP (Fig. S1, ESI†). 2-Pyridyldisulfide-terminated PNIPAM, i.e. SPDP capped PNIPAM, was prepared by an esterification reaction between amino-terminated PNIPAM and SPDP in PBS (0.01 M, pH 7.4). The structure of SPDP capped PNIPAM, which was synthesized by changing the PNIPAM–NH2–SPDP feed ratio to 1:2, dialyzed, and then freeze-dried, was confirmed by 1H NMR (Fig. S2, ESI†). The appearance of pyridine protons, i.e. 8.4 ppm, 7.8 ppm and 7.3 ppm, indicates the formation of PNIPAM–SPDP.
 |
| | Scheme 2 Synthesis of multi-sulfhydryl HSA, SPDP capped PNIPAM and multi-disulfide HSA–PNIPAM conjugates. | |
Synthesis of HSA–PNIPAM conjugates
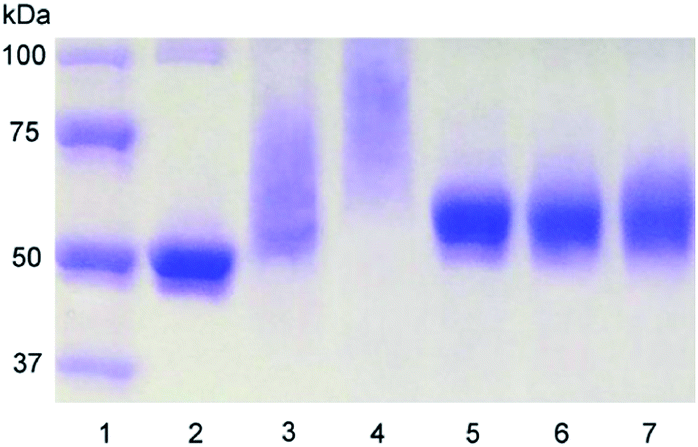
HSA–PNIPAM conjugates were prepared via conjugation of the 2-pyridyldisulfide group of PNIPAM–SPDP and the free sulfhydryl group of HSA (Scheme 2). The molecular structure of HSA–10-PNIPAM was examined using 1H NMR in D2O (Fig. 1) and confirmed by elemental analysis (Table. 1). HSA–10-PNIPAM conjugates in D2O, where the protons of pyridine were not visible compared to SPDP capped PNIPAM (Fig. S2, ESI†), indicates byproducts of pyridine 2-thione were removed during the purification procedure. During synthesis, a 2-pyridyldisulfide to sulfhydryl ratio of 5:1 mol/mol yielded an average of 5–11 PNIPAM chains grafted onto each HSA molecule. There were 11 polymer chains on each HSA–10-PNIPAM conjugate. It is reasonable to conclude that, after the HSA was reduced by TCEP, one intramolecular disulfide bond of native HSA was cleaved.69 Therefore, one or more polymer chains would couple HSA compare to the amount of SPDPs coupled HSA (Table 1). The HSA–5-PNIPAM and HSA–10-PNIPAM conjugates were analyzed by SDS-PAGE for a direct visual confirmation (Fig. 2). The conjugates appeared on the gel as higher-molecular-weight depigmentation, while the control HSA sample produced a band (Fig. 2, lane 2, 3, 4). With increasing numbers of chains, conjugates depigmentation migrated more slowly, i.e. more polymer chains slow gel migration because of their higher MW. After incubation of HSA–PNIPAM with TCEP, the depigmentation corresponding to the high-molecular-weight molecules almost disappeared, revealing bands corresponding to free HSA (Fig. 2, lane 5, 6, 7). This result confirmed that the polymer was conjugated to HSA via reducible bonds. It is also found that the MW of reduced HSA (Fig. 2, lane 5) is a little higher than that of unreduced HSA (Fig. 2, lane 2). Sodium dodecyl sulfate (SDS) is an anionic detergent used to denature proteins prior to gel electrophoresis. It is necessary to denature all proteins in a sample in order to separate them solely by their mass in a gel matrix. SDS efficiently breaks down secondary structures such as alpha helices and beta sheets (both primarily comprised of hydrogen bonds) as well as many tertiary structures;70 however, SDS does not break down any of the disulfide bonds that participate in many tertiary structures. Therefore, after treatment with TCEP and SDS, reduced HSA would seem like a linear chain. Unreduced HSA is globular and retains high gel mobility; unreduced conjugates should also be globular and migrate quickly, thus explaining the apparent discrepancy between the MW estimated by elemental analysis (HSA–5-PNIPAM (93937 Da) and HSA–10-PNIPAM (126937 Da)) and that determined by SDS-PAGE (Fig. 2, lanes 3, 4).
 |
| | Fig. 1 1H NMR spectra of HSA–10-PNIPAM in D2O. | |
Table 1 Synthesis and characterization of HSA–PNIPAM conjugates
| Sample |
Type of feed |
Yieldb (%) |
Ave. number of PNIPAM–NH2 chain in one conjugatec |
Hydrodynamic diameter at pH 7.4 (nm) |
LCST (°C) |
| Multi-sulfhydryl HSA, HSA coupled 5 or 10 SPDPs (feed ration) and reduced. Calculated based on the amount of feed HSA. Estimated using elemental analysis. |
| HSA |
— |
— |
— |
3.5 ± 0.2 |
— |
| HSA–5-PNIPAM |
HSA–5–SPDP–SHa/PNIPAM–SPDP |
71 |
5 |
5.48 ± 0.8 |
33.0 |
| HSA–10-PNIPAM |
HSA–10-SPDP–SHa/PNIPAM–SPDP |
75 |
11 |
14.3 ± 5.5 |
31.3 |
 |
| | Fig. 2 SDS-PAGE under non-reducing (2, 3, 4) and reducing (5, 6, 7) conditions. 1: marker; 2, 5: HSA; 3, 6: HSA–5-PNIPAM; 4, 7: HSA–10-PNIPAM. | |
Temperature-responsive behavior
PNIPAM is a well-known temperature-sensitive polymer.71 Above the LCST, the isopropyl side groups of PNIPAM gain entropy by releasing water molecules and becoming dehydrated. The hydrodynamic diameter of the conjugates was determined to be 5 and 14 nm by DLS (Table 1), and HSA–10-SPDP–SH did not increase in size (Fig. 3). In Table 1, LCST was defined as the temperature point at 10% of the maximum absorbance (Fig. S3, ESI†). Heating these solutions above the LCST to 37 °C increased the hydrodynamic diameter of HSA–5-PNIPAM and HSA–10-PNIPAM to 458 and 712 nm, respectively (Fig. 3), indicating that more PNIPAM chains on the protein surface result in increased hydrophobicity at temperatures above the LCST. Measured LCST were slightly higher for the HSA–PNIPAM conjugates than for the pure PNIPAM chains (ca. 30.2 °C by Fig. S3, ESI†). As expected, the LCST of the PNIPAM–protein conjugates was higher than that of PNIPAM owing to the increased hydrophilicity of the hybrid structure. As shown in (Fig. S3, ESI†), the LCST of the conjugates was slightly higher than that for PNIPAM and increased with decreasing numbers of PNIPAM chains conjugated to HSA. The uniformity of the phase-transition profiles suggested that the samples were not a mixture of free PNIPAM and HSA–PNIPAM conjugate. As the number of polymer chains conjugated to the protein decreases, its effects on the hydrophobic/hydrophilic balance of the hybrid structure changes. The effect of HSA on temperature responsiveness becomes dominant with increasing hydrophilicity of the hybrid. Thus, it is reasonable to assume that the LCST of the conjugates increases with decreasing numbers of conjugated PNIPAM chains.
 |
| | Fig. 3 Hydrodynamic size distributions as determined by DLS for HSA, HSA–10-SPDP–SH, HSA–5-PNIPAM, and HSA–10-PNIPAM at 25 °C or 37 °C. | |
Stability of the HSA conjugates against proteolytic degradation
Resistance of the HSA conjugates to enzyme degradation was assessed by incubation with chymotrypsin followed by SDS-PAGE. The proteins were released from the conjugates by TCEP reduction. Depigmentation of native HSA was observed after exposure to chymotrypsin (Fig. 4). The effect of PNIPAM versus HSA conjugates on reducing enzymatic protein hydrolysis was remarkable at 25 °C and 37 °C. Compared to native HSA, more HSA survives in the conjugates upon exposure to chymotrypsin, demonstrating the protective effect of the polymer. Moreover, conjugates with fewer PNIPAM chains (HSA–5-PNIPAM) were more weakly resistant to proteolytic attack than were conjugates with more polymer chains (HSA–10-PNIPAM). In addition, HSA depigmentation at 25 °C (Fig. 4, lane 2) was stronger than at 37 °C (Fig. 4, lane 5) after TCEP treatment under the same conditions, which is reasonable because higher temperature is favorable for chymotrypsin degradation of the protein. While, the conjugates treated with chymotrypsin at 25 °C yielded weaker HSA bands (Fig. 4, lane 3, 4) than those treated at 37 °C (Fig. 4, lane 6, 7), although slight enzymatic degradation was observed in the lower molecular weight bands. These results suggest that collapse of the polymer chains around HSA leads to more efficient protection of the protein.
 |
| | Fig. 4 SDS-PAGE of HSA–PNIAPM conjugates after incubation with chymotrypsin for 1.5 h at 25 °C (2, 3, 4) and 37 °C (5, 6, 7). The conjugates were reduced by TCEP prior to separation. 1: marker; 2, 5: HSA; 3, 6: HSA–5-PNIPAM; 4, 7: HSA–10-PNIPAM. | |
Binding affinity of warfarin to the HSA conjugates
HSA is composed of three homologous domains (I–III) and each domain has two subdomains (A and B). HSA is able to bind reversibly a large number of endogenous and exogenous compounds. The capability of warfarin depends largely on the existence of two major binding regions, namely Sudlow's site I and site II, which are located within specialized cavities in subdomains IIA and IIIA respectively.72,73 Warfarin, a widely used anticoagulant medication, binds with a high affinity to HSA.68 It is strongly fluorescent with excitation and emission maxima at 330 and 380 nm (approximately). Compared to free warfarin, the bound warfarin shows significantly higher emission intensity in aqueous solution.68 In this study, the emission spectra of warfarin–HSA in PBS (0.01 M, pH 7.4) showed an approximately 11-fold increase in emission intensity at 380 nm in comparison to free warfarin at the same concentration (Fig. S4, ESI†). Therefore, fluorescence intensity-based approaches are suitable for measuring warfarin–HSA interaction. There are several lysine residues around the binding site of HSA.74 Thus, it is might that the conjugation of PNIPAM to HSA decreased its binding affinity.
As shown in Fig. 5, under optimized conditions (37 °C, pH 7.4), the binding affinities of HSA–5-PNIPAM and HSA–10-PNIPAM were 91% and 78% that of the native protein, respectively. The results suggest that the polymer layer prevents warfarin penetration of the protein active site, for the number of polymer chains ranging between 5 and 11, and thus provides moderate protection against warfarin binding. The binding affinity of the conjugates is temperature-sensitive, where the conjugates exhibit enormous differences in warfarin binding affinity when the number of polymer chains was varied from 5 to 11. At 37 °C, HSA–5-PNIPAM exhibited 66% binding affinity and HSA–10-PNIPAM exhibited only 33% binding. The changes in binding affinity are largely attributed to the collapse of the polymer chain, which could efficiently prevent the approaching of warfarin molecules. We produced surface conjugates of 5–11 PNIPAM chains (5500 MW) grafted onto a protein molecule. It is expected shielding of the polymer chains on the HSA–10-PNIPAM would be more robust than molecules with fewer conjugates. Increasing the temperature to 37 °C induced a second conformational change in HSA–PNIPAM conjugates (Fig. S5c and d, ESI†) that may reduce binding affinity. The CD spectrum of HSA is typical of an α-helix structure.72 Conjugation of PNIPAM to HSA slightly reduced band intensity, suggesting PNIPAM decreased the helicity of the structure (Fig. S5a, ESI†). At different temperatures, the polymer–HSA interaction produced only mild changes in band intensity (Fig. S5b–d, ESI†), indicating that polymer collapse induces slight changes in the helical structure content of the protein. Besides, the binding affinity and CD of the recovered HSA were tested (Fig. S6, ESI†). The CD spectra show almost no difference between the recovered and the original protein, while slight decrease of warfarin binding affinity was observed. The reason could be that the modification of proteins via disulfide linkage is not completely reversible since the reduction agent such as TCEP would open a few disulfide bonds in the protein whenever it was cleaving the polymer chains.60 Polymer–protein coupling induces slight changes in secondary structure,75 although this does not significantly affect the native properties of the protein.76 As shown in Fig. S7 (ESI†), temperature had no effect on the fluorescence intensity of HSA–PNIPAM–NH2 and native HSA after warfarin binding. Therefore, the loss of conjugate binding affinity was mainly due to steric hindrance of the polymer; some loss might be due to the slight conformation shift.
 |
| | Fig. 5 Warfarin binding affinity of HSA–PNIPAM conjugates as a function of temperature. Relative warfarin fluorescence with HSA–PNIPAM conjugates at the indicated temperatures, reported relative to HSA. | |
The effect of thermal cycling on the warfarin binding affinity of HSA–PNIPAM conjugates
To investigate the effect of thermal cycling on the warfarin binding affinity of HSA–PNIPAM conjugates, the temperature was raised to 37 °C and maintained for 30 min. Although binding affinity decreased at 37 °C, it was almost fully recovered after recycling to 25 °C (Fig. 6a); warfarin binding affinity slightly decreased with thermal cycling (Fig. 6b). At 37 °C, PNIPAM is in a collapsed hydrophobic state; it rehydrates at 25 °C. The HSA–10-PNIPAM gives much lower warfarin binding affinity at 37 °C as shown in Fig. 6a, since the collapsed polymer chains is a key factor to prevent the protein from bonding the warfarin molecules. Under this condition the warfarin binding affinity would be mainly influenced by the number of the grafting polymers, related to the shielded area of the protein surface. Cyclic collapse and rehydration of the polymer chains create stresses on the HSA molecule. Thermal cycling through the LCST shifts the conjugates between loose coil and dense globule formations. The cyclic collapse and rehydration of the PNIPAM chains may create micro-environmental stresses that cause some HSA molecules to unfold, leading to conformational damage. This accounts for the slight shift in the CD spectrum (Fig. S5, ESI†).
 |
| | Fig. 6 Warfarin binding affinity of HSA–PNIPAM conjugates over 25 and 37 °C cycles. (a) Warfarin binding affinity of HSA–PNIPAM conjugates after one cycle. 25′ means one heating/cooling cycle and measured at 25 °C. (b) Warfarin binding affinity of HSA–PNIPAM conjugates after 7 cycle using cyclic temperatures were 25 and 37 °C. The dates were measured at 25 °C. | |
Conclusions
In this study, we developed a novel approach to graft controllable number of thermo-responsive polymer chains onto a protein through disulfide bond formation under mild condition. HSA–PNIPAM conjugates were synthesized and the influence of grafting number on warfarin binding affinity of the protein–polymer conjugates was examined. It is shown that conjugates with more polymers exhibited lower warfarin binding affinity since the polymers could efficiently prevent the approaching of chymotrypsin. Besides, the warfarin binding affinity could also be controlled by thermal cycling, and the inclusion of disulfide bonds permits dissociation under mild reducing conditions. The protein–polymer conjugates may be useful for drug delivery to a mildly reducing intracellular compartment such as in tumor cells.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant no. 51303105), the Natural Science Foundation of Guangdong Province (Grant no. S2013040014209), the China Postdoctoral Science Foundation (Grant no. 2013M540674), the Knowledge Innovation Program of Shenzhen Science and Technology Innovation Committee (Grant no. JCYJ20130401113535478) and the Stroke Screening and Prevention of Shenzhen Public Service Platform.
Notes and references
- C. Boyer, X. Huang, M. R. Whittaker, V. Bulmus and T. P. Davis, Soft Matter, 2011, 7, 1599 RSC
 .
. - A. S. Hoffman and P. S. Stayton, Prog. Polym. Sci., 2007, 32, 922 CrossRef CAS PubMed .
- E. S. Gil and S. M. Hudson, Prog. Polym. Sci., 2004, 29, 1173 CrossRef CAS PubMed .
- K. L. Heredia and H. D. Maynard, Org. Biomol. Chem., 2007, 5, 45 CAS .
- A. K. Shakya, H. Sami, A. Srivastava and A. Kumar, Prog. Polym. Sci., 2010, 35, 459 CrossRef CAS PubMed .
- H. A. Klok, Macromolecules, 2009, 42, 7990 CrossRef CAS .
- D. Pfister and M. Morbidelli, J. Controlled Release, 2014, 180, 134 CrossRef CAS PubMed .
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 459 CrossRef CAS .
- F. M. Veronese and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 453 CrossRef CAS .
- S. Hoffman, J. Controlled Release, 1987, 6, 297 CrossRef .
- S. G. Shoemaker, A. S. Hoffman and J. H. Priest, Appl. Biochem. Biotechnol., 1987, 15, 11 CrossRef CAS .
- N. Monji, A. S. Hoffman, J. H. Priest and R. L. Houghton, Thermally-induced phase separation immunoassay, US Pat., 4 780 409, 1988 .
- L. C. Dong and A. S. Hoffman, J. Controlled Release, 1986, 4, 223 CrossRef CAS .
- G. Chen and A. S. Hoffman, Bioconjugate Chem., 1993, 4, 509 CrossRef CAS .
- Z. L. Ding, R. B. Fong, C. J. Long, P. S. Stayton and A. S. Hoffman, Nature, 2001, 411, 59 CrossRef CAS PubMed .
- T. Shimoboji, Z. L. Ding, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 2001, 12, 314 CrossRef CAS PubMed .
- T. Shimoboji, E. Larenas, T. Fowler, S. Kulkarni, A. S. Hoffman and P. S. Stayton, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16592 CrossRef CAS PubMed .
- T. Shimoboji, E. Larenas, T. Fowler, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2003, 14, 517 CrossRef CAS PubMed .
- V. Bulmus, Z. L. Ding, C. J. Long, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 2000, 11, 78 CrossRef CAS PubMed .
- M. A. Gauthier and H. A. Klok, Chem. Commun., 2008, 2591 RSC .
- Z. L. Ding, C. J. Long, Y. Hayashi, E. V. Bulmus, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 1999, 10, 395 CrossRef CAS PubMed .
- M. Li, H. Li, P. De and B. S. Sumerlin, Macromol. Rapid Commun., 2011, 32, 354 CrossRef CAS PubMed .
- H. T. Ho, M. E. Levere, S. Pascual, V. Montembault, N. Casse, A. Caruso and L. Fontaine, Polym. Chem., 2013, 4, 675 RSC .
- H. Li, M. Li, X. Yu, A. P. Bapat and B. S. Sumerlin, Polym. Chem., 2011, 2, 1531 RSC .
- L. Tao, J. Liu, J. Xu and T. P. Davis, Org. Biomol. Chem., 2009, 7, 3481 CAS .
- L. Tao, J. Q. Liu and T. P. Davis, Biomacromolecules, 2009, 10, 2847 CrossRef CAS PubMed .
- J. Nicolas, G. Mantovani and D. M. Haddleton, Macromol. Rapid Commun., 2007, 28, 1083 CrossRef CAS .
- L. Tao, C. S. Kaddis, R. R. O. Loo, G. N. Grover, J. A. Loo and H. D. Maynard, Macromolecules, 2009, 42, 8028 CrossRef CAS PubMed .
- Z. L. Ding, G. H. Chen and A. S. Hoffman, Bioconjugate Chem., 1996, 7, 121 CrossRef CAS PubMed .
- V. Thilakarathne, V. A. Briand, Y. Zhou, R. M. Kasi and C. V. Kumar, Langmuir, 2011, 27, 7663 CrossRef CAS PubMed .
- C. Cummings, H. Murata, R. Koepsel and A. J. Russell, Biomacromolecules, 2014, 15, 763 CrossRef CAS PubMed .
- L. A. Canalle, D. W. P. M. Lowik and J. C. M. van Hest, Chem. Soc. Rev., 2010, 39, 329 RSC .
- G. Pound, J. M. McKenzie, R. F. M. Lange and B. Klumperman, Chem. Commun., 2008, 3193 RSC .
- S. Zalipsky, Bioconjugate Chem., 1995, 6, 150 CrossRef CAS .
- G. Pasut and F. M. Veronese, Prog. Polym. Sci., 2007, 32, 933 CrossRef CAS PubMed .
- B. S. Lele, H. Murata, K. Matyjaszewski and A. J. Russell, Biomacromolecules, 2005, 6, 3380 CrossRef CAS PubMed .
- L. Tao, G. Mantovani, F. Lecolley and D. M. Haddleton, J. Am. Chem. Soc., 2004, 126, 13220 CrossRef CAS PubMed .
- Z. Zarafshani, T. Obata and J. F. Lutz, Biomacromolecules, 2010, 11, 2130 CrossRef CAS PubMed .
- M. Yan, J. Ge, Z. Liu and P. Ouyang, J. Am. Chem. Soc., 2006, 128, 11008 CrossRef CAS PubMed .
- C. Cummings, H. Murata, R. Koepsel and A. J. Russell, Biomaterials, 2013, 34, 7437 CrossRef CAS PubMed .
- H. M. Li, A. P. Bapat, M. Li and B. S. Sumerlin, Polym. Chem., 2011, 2, 323 RSC .
- J. P. Magnusson, S. Bersani, S. Salmaso, C. Alexander and P. Caliceti, Bioconjugate Chem., 2010, 21, 671 CrossRef CAS PubMed .
- D. Ma, M. Li, A. J. Patil and S. Mann, Adv. Mater., 2004, 16, 1838 CrossRef CAS .
- Z. Yang, A. J. Mesiano, S. Venkatasubramanian, S. H. Gross, J. M. Harris and A. J. Russell, J. Am. Chem. Soc., 1995, 117, 4843 CrossRef CAS .
- M. Yan, Z. Liu, D. Lu and Z. Liu, Biomacromolecules, 2007, 8, 560 CrossRef CAS PubMed .
- M. Yan, J. Du, Z. Gu, M. Liang, Y. Hu, W. Zhang, S. Priceman, L. Wu, Z. H. Zhou, Z. Liu, T. Segura, Y. Tang and Y. Lu, Nat. Nanotechnol., 2010, 5, 48 CrossRef CAS PubMed .
- A. Valdebenito, P. Espinoza, E. A. Lissi and M. V. Encinas, Polymer, 2010, 51, 2503 CrossRef CAS PubMed .
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, Macromol. Rapid Commun., 2008, 29, 1172 CrossRef CAS .
- M. Li, P. De, H. M. Li and B. S. Sumerlin, Polym. Chem., 2010, 1, 854 RSC .
- L. Tao, J. Liu, J. Xu and T. P. Davis, Chem. Commun., 2009, 6560 RSC .
- N. W. S. Kam, Z. Liu and H. J. Dai, J. Am. Chem. Soc., 2005, 127, 12492 CrossRef CAS PubMed .
- S. Cerritelli, D. Velluto and J. A. Hubbell, Biomacromolecules, 2007, 8, 1966 CrossRef CAS PubMed .
- V. Bulmus, M. Woodward, L. Lin, N. Murthy, P. Stayton and A. Hoffman, J. Controlled Release, 2003, 93, 105 CrossRef CAS PubMed .
- Q. Geng, X. Sun, T. Gong and Z. R. Zhang, Bioconjugate Chem., 2012, 23, 1200 CrossRef CAS PubMed .
- G. Saito, J. A. Swanson and K. D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199 CrossRef CAS .
- S. Brocchini, S. Balan, A. Godwin, J. W. Choi, M. Zloh and S. Shaunak, Nat. Protoc., 2006, 1, 2241 CrossRef CAS PubMed .
- R. Cheng, F. Feng, F. H. Meng, C. Deng, J. Feijen and Z. Y. Zhong, J. Controlled Release, 2011, 152, 2 CrossRef CAS PubMed .
- E. Verheyen, S. van der Wal, H. Deschout, K. Braeckmans, S. de Smedt, A. Barendregt, W. E. Hennink and C. F. van Nostrum, J. Controlled Release, 2011, 156, 329 CrossRef CAS PubMed .
- J. Liu, V. Bulmus, D. L. Herlambang, C. Barner-Kowollik, M. H. Stenzel and T. P. Davis, Angew. Chem., Int. Ed., 2007, 46, 3099 CrossRef CAS PubMed .
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955 CrossRef CAS PubMed .
- M. S. Rosendahl, D. H. Doherty, D. J. Smith, S. J. Carlson, E. A. Chlipala and G. N. Cox, Bioconjugate Chem., 2005, 16, 200 CrossRef CAS PubMed .
- A. Basu, K. Yang, M. L. Wang, S. Liu, R. Chintala, T. Palm, H. Zhao, P. Peng, D. C. Wu, Z. F. Zhang, J. Hua, M. C. Hsieh, J. Zhou, G. Petti, X. G. Li, A. Janjua, M. Mendez, J. Liu, C. Longley, Z. Zhang, M. Mehlig, V. Borowski, M. Viswanathan and D. Filpula, Bioconjugate Chem., 2006, 17, 618 CrossRef CAS PubMed .
- D. C. Dieterich, A. J. Link, J. Graumann, D. A. Tirrell and E. M. Schuman, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9482 CrossRef CAS PubMed .
- A. Chilkoti, G. H. Chen, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 1994, 5, 504 CrossRef CAS .
- E. Bays, L. Tao, C. W. Chang and H. D. Maynard, Biomacromolecules, 2009, 10, 1777 CrossRef CAS PubMed .
- T. Shiroya, M. Yasui, K. Fujimoto and H. Kawaguchi, Colloids Surf., B, 1995, 4, 275 CrossRef CAS .
- J. Carlsson, H. Drevin and R. Axen, Biochem. J., 1978, 173, 723 CAS .
- M. Poor, Y. Li, S. Kunsagi-Mate, J. Petrik, S. Vladimir-Knezevic and T. Koszegi, J. Lumin., 2013, 142, 122 CrossRef CAS PubMed .
- W. E. Funk, H. Li, A. T. Iavarone, E. R. Williams, J. Riby and S. M. Rappaport, Anal. Biochem., 2010, 400, 61 CrossRef CAS PubMed .
- A. Rath, M. Glibowicka, V. G. Nadeau, G. Chen and C. M. Deber, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1760 CrossRef CAS PubMed .
- C. Boyer, V. Bulmus, J. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145 CrossRef CAS PubMed .
- J. N. Tian, J. Q. Liu, W. Y. He, Z. D. Hu, X. J. Yao and X. G. Chen, Biomacromolecules, 2004, 5, 1956 CrossRef CAS PubMed .
- H. Watanabe, U. Kragh-Hansen, S. Tanase, K. Nakajou, M. Mitarai, Y. Iwao, T. Maruyama and M. Otagiri, Biochem. J., 2001, 357, 269 CrossRef CAS .
- S. Curry, H. Mandelkow, P. Brick and N. Franks, Nat. Struct. Biol., 1998, 5, 827 CrossRef CAS PubMed .
- H. Tan, H. Jin, H. Mei, L. Zhu, W. Wei, Q. Wang, F. Liang, C. Zhang, J. Li, X. Qu, D. Shangguan, Y. Huang and Z. Yang, Soft Matter, 2012, 8, 2644 RSC .
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra06813k |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.