
Design and development of papain–urea loaded PVA nanofibers for wound debridement†
Abstract
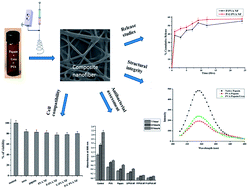
Devitalized tissues present in a wound bed serve as a reservoir for bacterial growth and contain elevated levels of inflammatory mediators that promote chronic inflammation and impair cellular migration necessary for wound repair. Effective wound cleansing and debridement are essential for granulation and re-epithelization. Among various debridement methods, enzymatic debridement is a highly selective method that uses naturally occurring proteolytic enzymes. Papain combined with urea has been widely used to remove necrotic/devitalized tissues. Our approach is to encapsulate papain and urea in PVA nanofibers to bring out sustained release to enable breakdown of fibrinous material in necrotic tissue and enhance wound healing. Physico-chemical characterization of nanofibers depicted the enzyme interaction with the polymer and also confirmed that the enzyme was evenly distributed in the nanofibers in an amorphous state. Fluorescence spectroscopy confirmed that the structural integrity of the enzyme was maintained after encapsulation. The results of antibacterial activity along with cell compatibility assays confirm the structural and functional integrity of the enzyme preparation along with the biocompatibility of the electrospun nanofiber and thereby provide more suitability as a dressing for wound debridement.
 Please wait while we load your content...
Please wait while we load your content...

