
Novel polymerisation of conducting thienothiophenes via vapour phase polymerisation: a comparative study†
M. P. Gustafson*a,
K. Matsumotob,
J. Janikowskic,
R. Kerrc,
D. R. MacFarlanea and
B. Winther-Jensenc
aARC Centre of Excellence for Electromaterials Science, Department of Chemistry, Monash University, Clayton, Victoria 3800, Australia. E-mail: Matthew.Gustafson@monash.edu
bDepartment of Chemistry and Biotechnology, Tottori University, Tottori 680-8552, Japan
cDepartment of Materials Engineering, Monash University, Clayton, Victoria 3800, Australia
First published on 24th October 2014
Abstract
Poly(thieno[3,2-b]thiophene) (PTT) and poly(dithieno[3,2-b:2′,3′-d]thiophene) (PDTT) were successfully synthesised via an oxidative vapour phase polymerisation (VPP) process and a typical electropolymerisation (EP) technique. This comparative study provides a characterisation of the conducting polymers (CPs) obtained. UV-Visible and Raman spectroscopy suggest the synthesis of PTT and PDTT via VPP yielded materials comparable to those produced via EP. This confirms VPP as a simple and straightforward alternative technique to achieve uniform thin-film polymers.
Since their discovery, conducting polymers (CPs) have found their way into a wide variety of electronic applications in research and commercial devices.1–10 Based upon abundant organic resources and housing a flexible, conjugated molecular structure, these materials can be cheaply manufactured and coated onto an array of surfaces.11–13 The latter is particularly advantageous, with future potential to coat existing surfaces and infrastructure with electronic sensors, circuitry and organic photovoltaics (OPVs).11,14
Thieno[3,2-b]thiophene (TT) and dithieno[3,2-b:2′,3′-d]thiophene (DTT) are widely studied and used in functional side-chains and co-polymers in light harvesting devices, such as in dye-sensitised solar cells and OPV heterojunction applications.15–20 When polymerised, the PTT and PDTT (Scheme 1) semiconducting materials possess the desired light absorption in the visible range, absorbing at approximately ∼500 nm. Furthermore, their stable, unique structure allows for manipulation of the conjugated backbone, thus influencing the band gap energy. This feature is particularly advantageous in a bulk heterojunction (BHJ) environment, providing the ability to ‘tune’ the band gap in order to optimise the material towards its application.21
 | ||
| Scheme 1 (a) Poly(thieno[3,2-b]thiophene) and (b) poly(dithieno[3,2-b:2′,3′-d]thiophene). | ||
Vapour Phase Polymerisation (VPP) is a technique originally developed by Mohammadi et al. and widely used by the CP community.22–25 The process for CP synthesis typically involves coating a substrate with an oxidant layer before placing into a heated chamber filled with pre-selected monomer vapours.23,25 At the appropriate temperature, these vapours undergo oxidative polymerisation on the surface of the oxidant.23,26 Additionally electrons are removed from the growing polymer chain by the oxidant, which becomes electrostatically bound to the polymer, acting as the dopant.23 VPP is able to produce extremely thin, uniform layers with a range of polymers and dopant ions.23,27,28 This technique is quite simple and effective when compared to other polymerisation techniques.
In this study, we successfully polymerised neat thieno[3,2-b]thiophene (TT) and dithieno[3,2-b:2′,3′-d]thiophene (DTT) using an oxidative VPP process using a Fe(III) p-toluenesulfonic acid (PTS) oxidant. Traditionally, these compounds are synthesised using a bromine substitution reaction involving functionalised monomers.29,30 Here we present a characterisation of these novel polymer films produced via the oxidative VPP process and compare their properties with PTT and PDTT synthesised via electropolymerisation (EP).
The thieno[3,2-b]thiophene (97%) (TT) was used as purchased from Sigma-Aldrich while dithieno[3,2-b:2′,3′-d]thiophene (DTT) was synthesised using an oxidative ring closure outlined by Jong and Janssen.31 Poly(thieno[3,2-b]thiophene) (PTT) and poly(dithieno[3,2-b:2′,3′-d]thiophene) (PDTT) were synthesised via VPP. This involved spin coating 0.5 mL of Fe(III)PTS (40% BuOH) oxidant onto a substrate at 1500 rpm for 30 s. The coated substrate was then placed into its respective monomer chamber and transferred into an oven at 70 °C for 30 min (PTT) or 5 h (PDTT). Upon retrieval, the polymer films were immersed overnight in EtOH (96% grade), to ensure complete removal of excess Fe(III)PTS oxidant.
The electropolymerisation of PTT and PDTT was based upon the cyclic voltammetry method described by Danieli, R. et al. and Corradini, A., et al.32,33 A three electrode setup employed a conducting substrate as a working electrode, platinum wire as a counter electrode and a Ag/AgCl leakless reference electrode submerged in a 0.1 M Bu4NClO4/DCM and 2 mM TT or 0.2 M Bu4NClO4/DCM and 2 mM DTT electrolyte. For an experimental description of characterisation methods conducted in this study see ESI S1.† Analysis of UV-Vis absorption spectra involved subtracting curves from a point at 1500 nm to improve a comparison, as light scattering resulted in high baselines. Unfortunately, the amorphous structure and insolubility of the polymer compounds excludes common characterisation techniques such as: X-ray diffraction, nuclear magnetic resonance, gas/liquid chromatography or mass spectroscopy.
The following data were gathered to explore the materials' optical and vibrational properties. These key characteristics assess the effectiveness of the CPs functionality in a light harvesting role.
The UV-Visible absorption spectra in Fig. 1 compare PTT and PDTT prepared via the two different polymerisation techniques. The two CP species present absorption spectra that differ between the methods. PTTEP appears less conjugated than PTTVPP given its lower π–π* transition at 493 nm compared to 506 nm.34 While PDTTEP demonstrates the opposite, displaying a longer wavelength and thus more conjugated π–π* transition compared to PDTTVPP. Additionally, the TT and DTT monomers absorb at 280 nm and 307 nm respectively (see ESI S2†).
 | ||
| Fig. 1 UV-Vis absorption spectra of PTT (a) and PDTT (b) polymerised via VPP (blue) and EP (red). Inset: π–π* transition (λmax) of each polymer. | ||
The absorption spectra were used to calculate the band gap energies (Eg) of the polymers to verify the ability to tailor a device via altering the band gap between PTT and PDTT. The spectra were plotted as (αhv)2 versus hv derived from eqn (1);
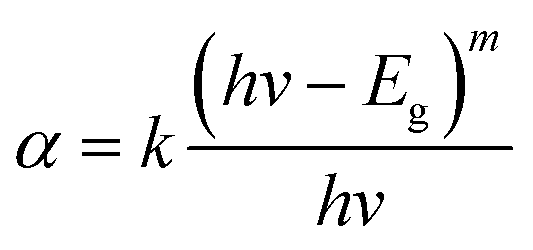 | (1) |
| PTT (eV) | PDTT (eV) | |
|---|---|---|
| VPP | 2.01 ± 0.02 | 1.93 ± 0.05 |
| EP | 1.97 ± 0.07 | 2.02 ± 0.03 |
Raman spectra (Fig. 2) compare polymerisation methods showing that the bond vibrations present in both sets of polymers are similar. This data supports the idea that PTT and PDTT synthesised via VPP contain the same bond formations and thus, comparable molecular structures to polymers synthesised via EP. This helps make VPP the favoured option when aiming to produce uniform, thin polymer films.
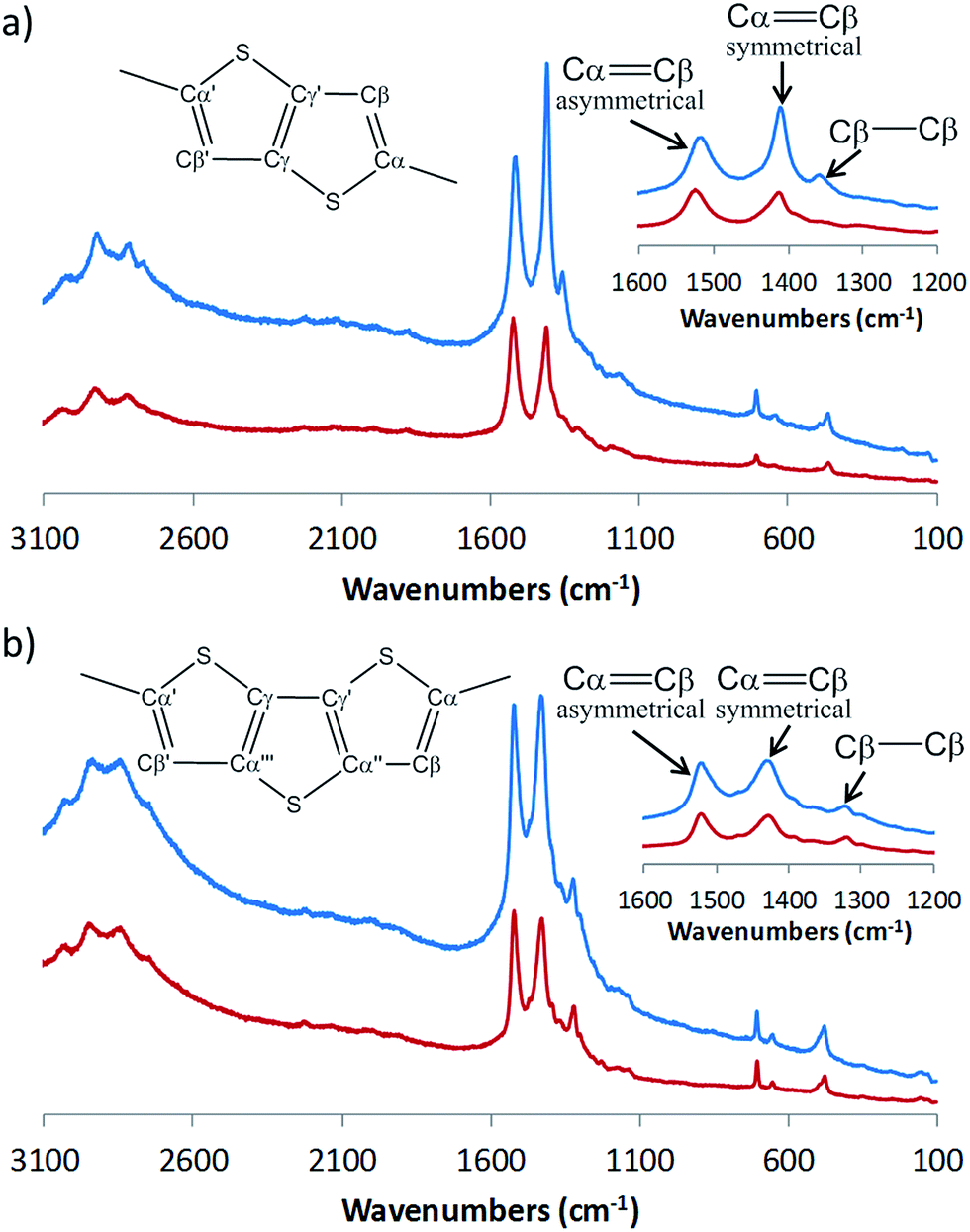 | ||
Fig. 2 Raman spectra of PTT (a) and PDTT (b) polymerised via VPP (blue) and EP (red). Inset: magnified spectra illustrating the difference in the Cα![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Cβ symmetrical stretch between PTT and PDTT. Cβ symmetrical stretch between PTT and PDTT. | ||
Step-wise chronoamperometry (CA) measurements were used to generate a linear relationship that followed Ohm's law (see ESI S3–S7†). From this, the conductivities of PTTVPP and PDTTVPP were calculated while conductivities for PTTEP and PDTTEP were obtained from the literature.32 The conductivities achieved by PTTVPP and PDTTVPP were 7.9 × 10−5 S cm−1 and 1.8 × 10−4 S cm−1 respectively. While the PTTEP and PDTTEP literature values are 3 × 10−6 S cm−1 and 3 × 10−3 S cm−1 respectively. In both techniques, PDTT is more conductive than PTT, suggesting this molecular architecture is preferential for charge transfer in PDTT films.
The cyclic voltammetry (CV) data shown in Fig. 3, compare the electrochemical profile of PTT and PDTT and their polymerisation methods. Due to the roughness of the Au substrates used, an accurate thickness could not be determined via standard or optical profilometry. From the baselines in Raman spectroscopy data, VPP and EP samples are thought to be approximately the same thickness. This is relevant as differences in film thickness would play a role in limiting diffusion of electrochemical species through the polymer. It is important to realise the different circumstances under which these polymers are synthesised. EP takes place in an electrolyte with the monomer being subjected to diffusion limitations as it migrates to the electrode surface. This process can create an irregular distribution of monomer available at the growth interface, resulting in rougher films especially at higher rates. However, Pringle et al., were able to produce smoother CP films via EP synthesis of PEDOT by running at a constant current rather than a CV or constant potential.37 Conversely, oxidative VPP uses an excess of monomer which condenses onto the oxidant surface faster than the polymerisation rate. This enables a constant, uniform monomer distribution which polymerises into the oxidant layer resulting in a smooth film.
 | ||
| Fig. 3 Cyclic voltammograms of (a) PTTVPP, (b) PDTTVPP, (c) PTTEP and (d) PDTTEP. All scans were run at 100 mV s−1 in 1 M LiClO4/PC and referenced with Pt vs. Fc+/Fc. | ||
The voltammograms show similar redox potentials (Table 2), with EP potentials comparable to those in the literature.33 The redox peaks from VPP samples are closer together than EP samples, indicating differences in material morphology which is supported by differences observed in imagery taken using scanning electron microscopy (SEM).
| PTT (V) | PDTT (V) | |
|---|---|---|
| VPP | 0.86 | 0.82 |
| EP | 0.62 | 0.71 |
The SEM imagery in Fig. 4, show the rough surface of the EP films, which is expected for electrodeposited CPs and acts as additional resistance.38,39 If surface dependent, the roughness would cause a delay in oxidation and reduction reactions, resulting in the peak separation and broadening seen in the EP samples in Fig. 3. Also, as PDTTEP appears to be rougher than PTTEP, this surface area increase helps explain the higher current densities achieved in PDTTEP.
 | ||
Fig. 4 SEM imagery of (a) PTTVPP, (b) PDTTVPP, (c) PTTEP and (d) PDTTEP at ×20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 magnification at 5 kV, scale bar = 1 μm. 000 magnification at 5 kV, scale bar = 1 μm. | ||
Conclusions
Thieno[3,2-b]thiophene and dithieno[3,2-b:2′,3′-d]thiophene were successfully polymerised via VPP achieving a comparable molecular structure to compounds prepared via electrodeposition. Although structurally similar, the EP and VPP techniques produce differing polymer morphologies, conjugation lengths and charge transfer capabilities. Films produced via VPP offer a simpler approach and a much smoother morphology compared to EP synthesis. From band gap calculations, thienothiophene compounds can allow for small variations in the molecular structure, adjusting the band structure to optimise the compound for a specific application.The results infer that although both techniques produce comparable materials, subtle differences from the deposition method allow for greater flexibility when designing a functional material.
Acknowledgements
The authors acknowledge funding from the Australian Research Council's Centers of Excellence scheme through the Australian Center of Excellence for Electromaterials Science and for use of facilities funded through the COE for Design in Light Metals within the Monash Centre for Electron Microscopy. DRM is also grateful to the ARC for funding through the Australian Laureate Fellowship scheme. The authors acknowledge the help received from Orawan Winther-Jensen for obtaining the SEM imagery for this study.Notes and references
- J. G. Hardy, J. Y. Lee and C. E. Schmidt, Curr. Opin. Biotechnol., 2013, 24, 847–854 CrossRef CAS PubMed.
- Y. Yajie, Z. Luning, L. Shibin, W. Zhiming, X. Jianhua, Y. Wenyao and J. Yadong, Nano-Micro Lett., 2013, 5, 40–46 CrossRef.
- M. Ates, Mater. Sci. Eng., C, 2013, 33, 1853–1859 CrossRef CAS PubMed.
- S. Selvakumar, N. Somanathan and K. A. Reddy, Propellants, Explos., Pyrotech., 2013, 38, 176–189 CrossRef CAS.
- U. Riaz, C. Nwaoha and S. M. Ashraf, Prog. Org. Coat., 2014, 77, 743–756 CrossRef CAS PubMed.
- R. Holze and Y. P. Wu, Electrochim. Acta, 2014, 122, 93–107 CrossRef CAS PubMed.
- G. Defieuw, R. Samijn, I. Hoogmartens, D. Vanderzande and J. Gelan, Synth. Met., 1993, 57, 3702–3706 CrossRef CAS.
- C. K. Chiang, C. R. Fincher, Y. W. Park, A. J. Heeger, H. Shirakawa, E. J. Louis, S. C. Gau and A. G. MacDiarmid, Phys. Rev. Lett., 1977, 39, 1098–1101 CrossRef CAS.
- C. K. Chiang, M. A. Druy, S. C. Gau, A. J. Heeger, E. J. Louis, A. G. MacDiarmid, Y. W. Park and H. Shirakawa, J. Am. Chem. Soc., 1978, 100, 1013–1015 CrossRef CAS.
- B. A. San Jose and K. Akagi, Polym. Chem., 2013, 4, 5144–5161 RSC.
- T. C. Huang, J. M. Yeh and C. Y. Lai, in Advances in Polymer Nanocomposites, ed. F. Gao, Woodhead Publishing, 2012, pp. 605–638 Search PubMed.
- C. J. Mulligan, M. Wilson, G. Bryant, B. Vaughan, X. Zhou, W. J. Belcher and P. C. Dastoor, Sol. Energy Mater. Sol. Cells, 2014, 120, 9–17 CrossRef CAS PubMed.
- M. Riede, B. Lüssem and K. Leo, in Comprehensive Semiconductor Science and Technology, ed. P. Bhattacharya, R. Fornari and H. Kamimura, Elsevier, Amsterdam, 2011, pp. 448–507 Search PubMed.
- J. C. Bijleveld, R. A. M. Verstrijden, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem., 2011, 21, 9224–9231 RSC.
- W. Tang, L. Ke, L. Tan, T. Lin, T. Kietzke and Z.-K. Chen, Macromolecules, 2007, 40, 6164–6171 CrossRef CAS.
- E. Nasybulin, M. Cox, I. Kymissis and K. Levon, Synth. Met., 2012, 162, 10–17 CrossRef CAS PubMed.
- S. Millefiorini, E. Kozma, M. Catellani and S. Luzzati, Thin Solid Films, 2008, 516, 7205–7208 CrossRef CAS PubMed.
- T.-H. Kwon, V. Armel, A. Nattestad, D. R. MacFarlane, U. Bach, S. J. Lind, K. C. Gordon, W. Tang, D. J. Jones and A. B. Holmes, J. Org. Chem., 2011, 76, 4088–4093 CrossRef CAS PubMed.
- S.-L. Li, K.-J. Jiang, K.-F. Shao and L.-M. Yang, Chem. Commun., 2006, 2792–2794 RSC.
- P. Morvillo, R. Diana, C. Fontanesi, R. Ricciardi, M. Lanzi, A. Mucci, F. Tassinari, L. Schenetti, C. Minarini and F. Parenti, Polym. Chem., 2014, 5, 2391–2400 RSC.
- A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed.
- A. Mohammadi, M. A. Hasan, B. Liedberg, I. Lundström and W. R. Salaneck, Synth. Met., 1986, 14, 189–197 CrossRef CAS.
- A. T. Lawal and G. G. Wallace, Talanta, 2014, 119, 133–143 CrossRef CAS PubMed.
- S. Shang, X. Yang, X.-m. Tao and S. S. Lam, Polym. Int., 2010, 59, 204–211 CAS.
- B. Winther-Jensen, J. Chen, K. West and G. Wallace, Macromolecules, 2004, 37, 5930–5935 CrossRef CAS.
- A. C. Fou and M. F. Rubner, Macromolecules, 1995, 28, 7115–7120 CrossRef CAS.
- Y. Fu, R. A. Weiss, P. P. Gan and M. D. Bessette, Polym. Eng. Sci., 1998, 38, 857–862 CAS.
- P. Subramanian, N. B. Clark, L. Spiccia, D. R. MacFarlane, B. Winther-Jensen and C. Forsyth, Synth. Met., 2008, 158, 704–711 CrossRef CAS PubMed.
- M. Heeney, C. Bailey, S. Tierney and I. McCulloch, Google Patents, US8114316B2, 2012.
- M. He, Google Patents, US7838623B2, 2010.
- M. J. Janssen and F. De Jong, J. Org. Chem., 1971, 36, 1645–1648 CrossRef CAS.
- R. Danieli, C. Taliani, R. Zamboni, G. Giro, M. Biserni, M. Mastragostino and A. Testoni, Synth. Met., 1986, 13, 325–328 CrossRef CAS.
- A. Corradini, M. Mastragostino, A. S. Panero, P. Prosperi and B. Scrosati, Synth. Met., 1987, 18, 625–630 CrossRef CAS.
- B. Kolodziejczyk, D. Mayevsky and B. Winther-Jensen, RSC Adv., 2013, 3, 4568–4573 RSC.
- A. Izgorodin, O. Winther-Jensen, B. Winther-Jensen and D. R. MacFarlane, Phys. Chem. Chem. Phys., 2009, 11, 8532–8537 RSC.
- N. M. Gasanly, I. Guler and K. Goksen, Cryst. Res. Technol., 2007, 42, 621–625 CrossRef CAS.
- J. M. Pringle, V. Armel and D. R. MacFarlane, Chem. Commun., 2010, 46, 5367–5369 RSC.
- T. Darmanin, M. Nicolas and F. Guittard, Phys. Chem. Chem. Phys., 2008, 10, 4322–4326 RSC.
- Z. Zhao-yang, T. Yi-jie, X. Xiao-qian, Z. Yong-jiang, C. Hai-feng and Z. Wen-wei, Synth. Met., 2012, 162, 2176–2181 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary information for novel polymerisation of conducting thienothiophenes via vapour phase polymerisation: a comparative study. See DOI: 10.1039/c4ra10200b |
| This journal is © The Royal Society of Chemistry 2014 |