
Effects of deposited ions on the photocatalytic activity of TiO2–Au nanospheres†
Abstract
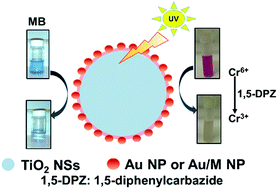
Photocatalytic TiO2–Au nanospheres (TiO2–Au NSs, 206 ± 23.7 nm) have been prepared and used as catalysts for the photo degradation of methylene blue (MB) and for the reduction of Cr6+ to Cr3+. TiO2 NSs are firstly prepared from titanium isopropoxide (TIP) via a solvothermal method. The TiO2 NSs are then sequentially modified with poly-(sodium-4-styreneulfonate) (PSS) and poly(diallyldimethylammonium chloride) (PDADMAC), which then interact with Au NPs (15 ± 1.3 nm) as seeds. Through reduction of HAuCl4 by ascorbic acid, core–shell structures of TiO2–Au NSs are prepared. Under UV irradiation, TiO2–Au NSs provide highly catalytic activity for the degradation of MB and reduction of Cr6+ within 15 and 60 min, respectively. The TiO2–Au NSs relative to commercial TiO2 (P25) and TiO2 NSs provide 1.8 and 1.2-fold activity higher for the photo degradation of MB, and 4.3 and 1.8-fold higher for the reduction of Cr6+. TiO2–Au/Hg and TiO2–Au/Ag NSs that are prepared from the deposition of Hg2+ and Ag+ onto TiO2–Au NSs, respectively, allow degradation of MB within 10 min, with activities 4.2- and 3.3-fold greater than that of the TiO2–Au NSs. The present study reveals that TiO2–Au/Hg and TiO2–Au/Ag NSs are effective for removal of organic pollutants, while TiO2–Au NSs are useful for the reduction of Cr6+.
 Please wait while we load your content...
Please wait while we load your content...

