
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A photochromic and thermochromic fluorescent protein†
Y.
Shen
,
M. D.
Wiens
and
R. E.
Campbell
*
Department of Chemistry, University of Alberta, Edmonton, Alberta T6G 2G2, Canada. E-mail: robert.e.campbell@ualberta.ca
First published on 21st October 2014
Abstract
We report a photochromic and thermochromic fluorescent protein that exhibits a reversible and striking visible colour switch between yellow and red. The protein has been characterized in terms of its light, temperature and pH-dependence. Based on a mutational analysis we propose that the colour switch mechanism involves chromophore protonation coupled with E–Z isomerization.
Photochromism is a change of colour induced by irradiation with light, while thermochromism is a change of colour due to a change of temperature.1 Both photochromism and thermochromism have been observed in inorganic and organic compounds, however thermochromism is much less common in biological systems.1b,2 One mechanism by which molecular photochromism can occur is a photo-induced isomerization to a second stable state with a different absorption wavelength. For such molecules, the rate of conversion is accelerated with increased temperature, and therefore the photochromic and thermochromic properties are intimately coupled.1b,3,4
Only a handful of classes of naturally occurring photochromic proteins have been identified. Representative classes of photochromic proteins include bacteriophytochromes,5 rhodopsins,6 and fluorescent proteins (FPs).7 The first photochromic FP to be described was the reversible ‘kindling’ protein, asFP595, from the sea anemone Anemonia sulcata.7 Reports of a number of other reversibly photochromic FPs followed, including many with faster switching properties or red-shifted emission colour, including: Dronpa,8 rsFastlime,9 mTFP0.7,10 rsCherry,11 and rsTagRFP.12 Mechanistic investigations of photochromic proteins have revealed that the photoswitching mechanism is typically based on coupled protonation changes and E–Z isomerizations of the protein chromophores.9,11,13,14
Although the photochromism of FPs has been studied extensively, far fewer examples of FP thermochromism have been reported.15 Here we report an engineered FP that exhibits reversible visible photochromism and thermochromism and is exquisitely sensitive to physiologically relevant changes in both pH and temperature.
We serendipitously discovered this photochromic and thermochromic FP during the engineering of a long Stokes shift (LSS) variant of mApple.16 mApple is a monomeric red fluorescent protein engineered from Discosoma sp. red FP (DsRed).17 LSS fluorescence is enabled by the introduction of an excited state proton transfer (ESPT) pathway into the protein. Accordingly, in LSSm Apple, blue light excitation of the neutral (protonated) chromophore leads to deprotonation and the formation of the excited state anionic chromophore that emits red light.18 Screening of a library of randomly mutated variants of an intermediate template (mApple-W143L/I161S/K163E, numbered according to DsRed's sequence), led to the identification of a bright variant that was picked for further characterization. The purified protein appeared yellow due to the blue light absorbing neutral chromophore. However, when this protein solution was left on ice for a few minutes under ambient light, it was observed to turn a bright magenta colour. Removing the protein from the ice caused a conversion back to the yellow state, revealing its intriguing reversible thermochromic property. DNA sequencing revealed that this protein contained the two additional mutations S146T and R164W (Fig. S1†). The hydroxyl moiety of 146 directly interacts with the phenol/phenolate group of the chromophore, while residue 164 is on the surface of the FP with its side chain directed towards the solvent. The T146S reversion mutation abolished the photochromic and thermochromic character of the protein. Reversion of the mutations at 164 produced a protein that still exhibited chromism. This result clearly demonstrated the importance of T146 for the photo- and thermochromism. This new variant was designated as switchable hypersensitive red FP (shyRFP).
ShyRFP, similar to earlier generations of its mApple template16 and the mApple derived genetically encoded biosensor R-GECO1,19 shows a photoinduced change in spectrum. However, unlike these previous examples where the change is of little visible consequence, shyRFP exhibits a dramatic visible colour shift from a yellow state (lower energy state) to a red state (higher energy state) upon irradiation with violet or blue light at room temperature (Fig. 1A). A similar colour change also occurs when the temperature of the protein solution is decreased to 0 °C under normal indoor illumination. When maintained in the dark, the protein thermally reverts from the red state back to the yellow state (Fig. 1B). Absorbance (Fig. 1C) and fluorescence spectral measurements (Fig. 1D) revealed that the yellow state is associated with a single absorbance peak at 450 nm. Excitation at this peak leads to LSS red fluorescence with a peak emission at 596 nm. In contrast, the red state, produced by illumination of the protein solution with a violet laser (405 nm) or blue light (470/40 nm), was associated with a second species with maximal absorbance at 545 nm (Fig. 1C). No significant red fluorescence emission was observed when this longer wavelength species was excited (Fig. 1D). After converting to the red state, both the absorbance and fluorescence of the 450 nm absorbing species decreased to ∼60% of the intensity in the original yellow state. The protein remained monomeric in both the yellow and red states (Fig. S2†), indicating the photochromism of this protein does not involve protein oligomerization through β-barrel structure rearrangement, as observed for Dronpa.20
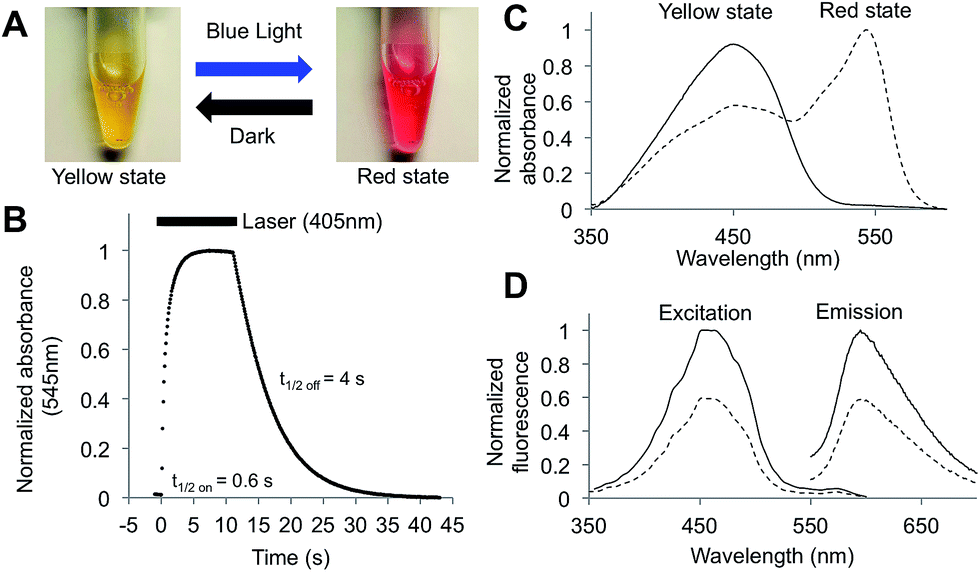 | ||
| Fig. 1 Photochromic properties of shyRFP. (A) Visible colour appearance of purified protein in the yellow and red states. (B) Absorbance (545 nm) change upon violet light (405 nm) activation and dark conversion (22 °C, pH = 7.5). Bar above the curve indicates the time when laser illumination is on. (C) Photochromic absorbance spectrum change. (D) Photochromic fluorescence spectrum change (yellow state, solid line; red state, dashed line). Note that the 545 nm absorbing species is not apparent in the red state excitation spectrum. | ||
To further characterize the photochromic properties of shyRFP, the kinetics of the photoswitching and thermal recovery were measured and fit as first order reactions. When illuminated by a violet laser (405 nm, 150 mW) at room temperature and a pH of 7.5, the protein quickly converts to the red state with an activation half-time of 0.6 s. Thermal reversion is slower than the activation with a half-time of 4 s (Fig. 1B). The photoactivation process showed strong light intensity dependence, with both the reaction rate and relative change of absorbance at 545 nm increasing as light intensity increased (Fig. S3†). The light-dependent thermochromism was characterized by measuring the absorbance spectra of the red state (produced using the 405 nm laser) at different temperature values ranging from 6 to 42 °C (Fig. 2A). As the temperature decreases from 42 to 6 °C, the absorbance of 450 nm decreased by 20%, while the 545 nm absorbance dramatically increased 3-fold resulting in a red solution (Fig. 2B). This result corresponded well to the observation of red colour protein solution after ambient room light illumination on ice. This dramatic shift in absorbance and visible colour can be explained by the difference in extinction coefficients for the yellow (30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) and red states (142000 M−1 cm−1). This means that, even with a modest degree of conversion of the yellow state to the red state, the solution will appear magenta. The absorbance peak ratio exhibited the largest changes in the 30 to 42 °C range (Fig. 2C), thus making this protein a possible candidate for a molecular thermometer in mammalian cells or tissues.
000 M−1 cm−1) and red states (142000 M−1 cm−1). This means that, even with a modest degree of conversion of the yellow state to the red state, the solution will appear magenta. The absorbance peak ratio exhibited the largest changes in the 30 to 42 °C range (Fig. 2C), thus making this protein a possible candidate for a molecular thermometer in mammalian cells or tissues.
 | ||
| Fig. 2 Thermochromic properties of shyRFP. (A) Absorbance spectra of laser photoswitched protein at 6 and 42 °C at pH 7.5. (B) Change of peak absorbance (545 nm, hollow circle; 450 nm, triangle). (C) Change of peak absorbance ratio (545 nm/450 nm) from 6 °C to 42 °C under violet light (405 nm) illumination (pH = 7.5). (D) Decay kinetics of absorbance at 545 nm under various temperatures at pH 7.5. (E) Arrhenius plot of thermal decay kinetics. | ||
The dark recovery process was strongly dependent on temperature. Specifically, as the temperature increased, the half-time of reversion decreased (Fig. 2D). Using the Arrhenius plot (Fig. 2E), the activation energy of the thermal relaxation was calculated as 34 kJ mol−1, which is comparable with other organic photochromic and thermochromic systems such as spiropyrans (9 to 27 kJ mol−1).21 This relatively low energy barrier explains the short half time of thermal relaxation for shyRFP (4 s), relative to almost all other photochromic FPs including Dronpa (840 min),9 mTFP0.7 (4 min),10 rsTagRFP (65 min)12 and rsCherry (40 s).11
To investigate the influence of the protein chromophore protonation state on the photo- and thermochromism, kinetic measurements of photoactivation (Fig. 3A and B) and thermal recovery (Fig. 3C and D) were performed at various pH values between 3 and 11. These measurements revealed that both processes were accelerated at lower pH values, suggesting that the mechanism of photochromism was associated with the chromophore protonation state. Absorbance (Fig. 3A and B) and fluorescence spectra (Fig. S4†) of the yellow state protein at various pH values were measured. At low pH (3 to 6), the absorbance and fluorescence spectra revealed the coexistence of three distinct states of chromophore: a blue-absorbing/red-emitting LSS species, a yellow-absorbing/non-fluorescent species, and an orange-absorbing/red-emitting species (Fig. S5†). At neutral to high pH (7 to 10), the chromophore was primarily the blue-absorbing red-emitting LSS species. Higher pH values resulted in denaturation of the protein. The fact that the absorbance spectrum, at any pH, is not identical to the photoactivated form suggested to us that a simple equilibrium between the protonated and deprotonated forms of the chromophore is insufficient to explain the chromic properties of shyRFP.
 | ||
| Fig. 3 pH dependence of shyRFP. (A and B) Photoactivation traces and rates at different pH values. (C and D) Thermal recovery traces and rates at different pH values. (E and F) Normalized absorbance spectra and absorbance ratio (550 nm/450 nm) in the yellow state under various pH values. | ||
To further explore the mechanism of chromism in shyRFP, saturation mutagenesis was conducted at residues in close proximity to the chromophore (i.e., residues 146, 161 and 163). Mutations at residue 146 resulted in significantly reduced (T146C and T146G) or complete loss (T146S) of photochromism. However, mutations at 146 did not alter the LSS spectral character, which indicates that residue 146 is not involved in the ESPT pathway. At residue 161 and 163, several mutations (S161A, S161F, E163I and E163V) resulted in the disappearance of the blue absorbing/LSS fluorescent species and yielded regular Stokes shift red FP type variants with no chromic changes. This result indicates that residues 161 and 163 are essential for the ESPT pathway18 that gives rise to the LSS fluorescence. In addition, considering the relative positions of residues 161 and 163 to the protein chromophore (Fig. 4), it appears that the LSS species, associated with the yellow state of shyRFP, is most likely in the E conformation.18,22
 | ||
| Fig. 4 Proposed mechanism of shyRFP photochromism and thermochromism. (A) Proposed schematic of the chromophore states involved in conversion. (B) Proposed isomers of the chromophore and the important amino acids in the chormophore environment. | ||
The photophysical characterization and site-directed mutagenesis results with shyRFP led to the proposal of a mechanism that accounts for the key features of this photochromic and thermochromic phenomenon (Fig. 4A). For the photochromism, violet/blue light illumination changes the chromophore from a 450 nm absorbing LSS fluorescent form (A/A* form) to a 545 nm absorbing non-fluorescent form (R form), which we conclude is anionic due to its long wavelength absorbance at 545 nm. In the dark, the 545 nm absorbing species R thermally reverts back to its neutral ground state, A form. The ground state equilibrium between A and B shifts towards the red fluorescent B form at low pH due to a “reverse protonation” effect that has been reported for LSS RFP mKeima.23 At low pH the side chain of E163 is protonated and cannot stabilize the hydrogen bond network with the neutral chromophore. Accordingly, the anionic B form of the chromophore is favoured (Fig. 4B).
The non-fluorescent R form has a blue-shifted absorbance maximum (545 nm) relative to the red fluorescent B form (580 nm). The fact that these two states are not identical at any pH rules out the possibility that violet/blue light illumination simply reversibly shifts the equilibrium between the protonated A state and deprotonated B state of the protein chromophore. Yet the reaction rate is highly dependent on the pH, indicating that protonation/deprotonation is coupled with chromism. Accordingly, in addition to a change in chromophore protonation state, the mechanism of the photochromism must also involve an alternate structure, conformation, or microenvironment for the illuminated form. Our currently preferred explanation is that violet/blue light illumination induces an E–Z isomerization of the chromophore. Our mutational study suggested that the LSS fluorescent A form (yellow state) is in the E conformation, and that the photoswitching process changes the chromophore conformation to the Z isomer, which is stable in the anionic state (Fig. 4B). In the dark, the Z isomer (red state) is able to thermally relax back to the E conformation (yellow state) at room temperature due to the low activation energy. In short, the photochromism and thermochromism of shyRFP are closely coupled, with illumination pushing the chromophore towards the red R state (anionic Z isomer) and the thermal relaxation pushing the chromophore towards the yellow A state (protonated E isomer).
Due to its complex photophysics and rapid photoswitching behaviour, shyRFP is an unlikely candidate for applications in conventional fluorescence microscopy.24 However, shyRFP could potentially find application in some specialized imaging experiments. For example, temperature and pH measurements based on the absorbance peak ratios or the kinetics of dark reversion for photoactivated shyRFP might be accessible to absorption microscopy. As both of these variables are inherently coupled, care must be taken to extract the desired information. With its large and distinct reversible absorbance spectra shift, shyRFP could also serve as a model for photoswitching studies and provide insight into the photoactivation mechanism of mApple variants. ShyRFP is the first example of an LSS RFP with a reversible fluorescence change; therefore, like Dronpa, rsCherry, and rsTagRFP, it could in principle be applied for super-resolution microscopy techniques, such as PALM or RESOLFT25 Furthermore, the shyRFP photoconversion is accompanied by significant absorbance changes that potentially allows its application in photochromic FRET.12,26 ShyRFP also has considerable potential as a teaching tool as it visually changes colour without expensive equipment and reacts strongly to light, temperature, and pH. ShyRFP could also be used in more frivolous pursuits such as transgenic plants that change colour in response to temperature changes. We are certain that further development and evolution of shyRFP will lead to more useful additions to the FP toolbox for use in live cell imaging.
Acknowledgements
This research was supported by the Natural Sciences and Engineering Research Council of Canada, the University of Alberta (Queen Elizabeth II scholarship to MDW), and Alberta Innovates (scholarship to YS). We thank Dr Serpe and Dr Gibbs-Davis for kindly providing access to the equipment, and Jiahui Wu and Nazanin Assempour for technical assistance.Notes and references
- (a) J. H. Day, Chem. Rev., 1963, 63, 65 CrossRef CAS; (b) H. Dürr and H. Bouas-Laurent, Photochromism: Molecules and Systems: Molecules and Systems, Gulf Professional Publishing, 2003 Search PubMed.
- A. Seeboth, D. Lotzsch, R. Ruhmann and O. Muehling, Chem. Rev., 2014, 114, 3037 CrossRef CAS PubMed.
- G. Berkovic, V. Krongauz and V. Weiss, Chem. Rev., 2000, 100, 1741 CrossRef CAS PubMed.
- M. Maafi, Molecules, 2008, 13, 2260 CrossRef CAS PubMed.
- S. H. Bhoo, S. J. Davis, J. Walker, B. Karniol and R. D. Vierstra, Nature, 2001, 414, 776 CrossRef CAS PubMed.
- N. Hampp, Chem. Rev., 2000, 100, 1755 CrossRef CAS PubMed.
- D. M. Chudakov, V. V. Belousov, A. G. Zaraisky, V. V. Novoselov, D. B. Staroverov, D. B. Zorov, S. Lukyanov and K. A. Lukyanov, Nat. Biotechnol., 2003, 21, 191 CrossRef CAS PubMed.
- R. Ando, H. Mizuno and A. Miyawaki, Science, 2004, 306, 1370 CrossRef CAS PubMed.
- A. C. Stiel, S. Trowitzsch, G. Weber, M. Andresen, C. Eggeling, S. W. Hell, S. Jakobs and M. C. Wahl, Biochem. J., 2007, 402, 35 CrossRef CAS PubMed.
- N. J. Henderson, H.-w. Ai, R. E. Campbell and J. S. Remington, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6672 CrossRef PubMed.
- A. C. Stiel, M. Andresen, H. Bock, M. Hilbert, J. Schilde, A. Schönle, C. Eggeling, A. Egner, S. W. Hell and S. Jakobs, Biophys. J., 2008, 95, 2989 CrossRef CAS PubMed.
- F. V. Subach, L. Zhang, T. W. Gadella, N. G. Gurskaya, K. A. Lukyanov and V. V. Verkhusha, Chem. Biol., 2010, 17, 745 CrossRef CAS PubMed.
- M. Andresen, M. C. Wahl, A. C. Stiel, F. Gräter, L. V. Schäfer, S. Trowitzsch, G. Weber, C. Eggeling, H. Grubmüller, S. W. Hell and S. Jakobs, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13070 CrossRef CAS PubMed.
- (a) S. Habuchi, P. Dedecker, J. Hotta, C. Flors, R. Ando, H. Mizuno, A. Miyawaki and J. Hofkens, Photochem. Photobiol. Sci., 2006, 5, 567 RSC; (b) S. Pletnev, F. V. Subach, Z. Dauter, A. Wlodawer and V. V. Verkhusha, J. Mol. Biol., 2012, 417, 144 CrossRef CAS PubMed; (c) S. Gayda, K. Nienhaus and G. U. Nienhaus, Biophys. J., 2012, 103, 2521 CrossRef CAS PubMed.
- (a) P. Leiderman, D. Huppert and N. Agmon, Biophys. J., 2006, 90, 1009 CrossRef CAS PubMed; (b) A. R. Faro, P. Carpentier, G. Jonasson, G. Pompidor, D. Arcizet, I. Demachy and D. Bourgeois, J. Am. Chem. Soc., 2011, 133, 16362 CrossRef PubMed; (c) K. S. Sarkisyan, I. V. Yampolsky, K. M. Solntsev, S. A. Lukyanov, K. A. Lukyanov and A. S. Mishin, Sci. Rep., 2012, 2, 608 Search PubMed.
- N. C. Shaner, M. Z. Lin, M. R. McKeown, P. A. Steinbach, K. L. Hazelwood, M. W. Davidson and R. Y. Tsien, Nat. Methods, 2008, 5, 545 CrossRef CAS PubMed.
- M. V. Matz, A. F. Fradkov, Y. A. Labas, A. P. Savitsky, A. G. Zaraisky, M. L. Markelov and S. A. Lukyanov, Nat. Biotechnol., 1999, 17, 969 CrossRef CAS PubMed.
- K. D. Piatkevich, V. N. Malashkevich, S. C. Almo and V. V. Verkhusha, J. Am. Chem. Soc., 2010, 132, 10762 CrossRef CAS PubMed.
- J. Wu, L. Liu, T. Matsuda, Y. Zhao, A. Rebane, M. Drobizhev, Y. F. Chang, S. Araki, Y. Arai, K. March, T. E. Hughes, K. Sagou, T. Miyata, T. Nagai, W. H. Li and R. E. Campbell, ACS Chem. Neurosci., 2013, 4, 963 CrossRef CAS PubMed.
- H. Mizuno, T. K. Mal, M. Walchli, A. Kikuchi, T. Fukano, R. Ando, J. Jeyakanthan, J. Taka, Y. Shiro, M. Ikura and A. Miyawaki, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9227 CrossRef CAS PubMed.
- K. K. Kalninsh, J. Struct. Chem., 1998, 39, 642 CrossRef CAS.
- D. M. Shcherbakova, M. A. Hink, L. Joosen, T. W. J. Gadella and V. V. Verkhusha, J. Am. Chem. Soc., 2012, 134, 7913 CrossRef CAS PubMed.
- (a) T. Kogure, S. Karasawa, T. Araki, K. Saito, M. Kinjo and A. Miyawaki, Nat. Biotechnol., 2006, 24, 577 CrossRef CAS PubMed; (b) S. Violot, P. Carpentier, L. Blanchoin and D. Bourgeois, J. Am. Chem. Soc., 2009, 131, 10356 CrossRef CAS PubMed.
- (a) M. W. Davidson and R. E. Campbell, Nat. Methods, 2009, 6, 713 CrossRef CAS; (b) D. M. Chudakov, M. V. Matz, S. Lukyanov and K. A. Lukyanov, Physiol. Rev., 2010, 90, 1103 CrossRef CAS PubMed.
- M. Fernandez-Suarez and A. Y. Ting, Nat. Rev. Mol. Cell Biol., 2008, 9, 929 CrossRef CAS PubMed.
- C. Don Paul, C. Kiss, D. A. Traore, L. Gong, M. C. Wilce, R. J. Devenish, A. Bradbury and M. Prescott, PLoS One, 2013, 8, e75835 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Material and methods, sequence information of shyRFP, and other supplementary figures. See DOI: 10.1039/c4ra10107c |
| This journal is © The Royal Society of Chemistry 2014 |