
Benzylamine as an arylcarboxy surrogate: a copper catalysed o-benzoxylation of 2-phenylpyridines using benzyl amines†
Abstract
Different reactivities and selectivities of Cu and Pd catalysts have been demonstrated in the reactions of benzylamines with 2-phenylpyridines. Although Pd is reported to give o-arylation (ArCO–), Cu introduces an arylcarboxy group (ArCOO–) at the proximal site of the directing group. For the first time benzylamine has been utilised as a synthetic equivalent of an arylcarboxy group.
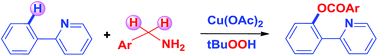
 Please wait while we load your content...
Please wait while we load your content...

