
New CHARMM force field parameters for dehydrated amino acid residues, the key to lantibiotic molecular dynamics simulations†
Eleanor R. Turpin‡
*a,
Sam Mulhollandab,
Andrew M. Tealeb,
Boyan B. Boneva and
Jonathan D. Hirstb
aSchool of Life Sciences, University of Nottingham, UK. E-mail: eleanor.turpin@nottingham.ac.uk
bSchool of Chemistry, University of Nottingham, UK
First published on 22nd September 2014
Abstract
Lantibiotics are an important class of naturally occurring antimicrobial peptides containing unusual dehydrated amino acid residues. In order to enable molecular dynamics simulations of lantibiotics, we have developed empirical force field parameters for dehydroalanine and dehydrobutyrine, which are compatible with the CHARMM all-atom force field. The parameters reproduce the geometries and energy barriers from MP2/6-31G*//MP2/cc-pVTZ quantum chemistry calculations. Experimental, predicted and calculated NMR chemical shifts for the amino protons and α-, β- and carbonyl carbon atoms of the dehydrated residues are consistent with a significant charge redistribution. The new parameters are used to perform the first molecular dynamics simulations of nisin, a widely used but poorly understood lantibiotic, in an aqueous environment and in a phospholipid bilayer. The simulations show surface association of the peptide with membranes in agreement with solid state NMR data and formation of β-turns in agreement with solution NMR.
Introduction
Bacterial adaptations and resistance to antibiotics present an increasing challenge to healthcare in hospitals and in the community, as well as in maintaining a safe and nutritious food supply for an increasing global population. Peptidoglycan-targeting antimicrobial peptides remain underutilized in bacterial management and resistance remains uncommon. Used alone or as facilitators in combination with other antimicrobials, these compounds hold much promise and offer a model for the development of novel molecules for bacterial control.Lanthionine antibiotics, or lantibiotics, are a group of antimicrobial peptides that are highly efficient against Gram-positive bacteria, have good environmental stability and have no known toxicity to humans.1–3 They are characterized by macrocyclic rings closed by unusual thioether bonds, formed by joining cysteine thiols to dehydroalanine (Dha) and dehydrobutyrine (Dhb), derived by post-translational dehydration of serine and threonine, respectively. This broad category of antibiotics is classified into three groups: class I lantibiotics are modified by separate dehydratases and cyclases; class II are modified by a single enzyme and class III have little antibiotic activity and perform other functions.1 There are over 80 novel lantibiotics, many discovered since 1985,4 and this continues to be a growing area for pharmaceutical exploitation.
The best understood and most widely used class I lantibiotic, nisin, is effective against Staphylococci, Clostridia and Lysteria and has numerous applications in food protection worldwide. Nisin targets pyrophosphorylated peptidoglycan intermediates lipid II and undecaprenyl pyrophosphate (11PP), present in the outer leaflet of bacterial membranes. It forms membrane-lytic complexes with lipid II, which dissipate transmembrane potentials and solute gradients.5,6 Membrane complexes with 11PP are inhibitory to cell wall biosynthesis.5,7 Nisin also deregulates bacterial division and cell wall morphogenesis8 and can inhibit spore outgrowth.9,10
Nisin is produced ribosomally in Lactococcus lactis and modified to have a structure that includes five thioether-bonded macrocyclic rings and three dehydrated amino residues – Dhb2, Dha5 and Dha33 (Fig. 1). Dha5 is essential to the spore outgrowth activity of nisin.9 The mature peptide is positively charged and readily binds negative and neutral membranes.11,12 NMR studies of nisin in solution13–16 and membrane-mimetic detergent micelles17 have shown that rings A, B and C are joined to the inter-linking rings D and E by a flexible region, and that the linear tails at the N- and C-termini are also flexible.18 Disulfide-linked macrocyclic peptide analogs of the nisin target-recognition motif 1–12 are conformationally similar to the natural peptide but can form in the absence of the nisB/C enzymatic machinery.19
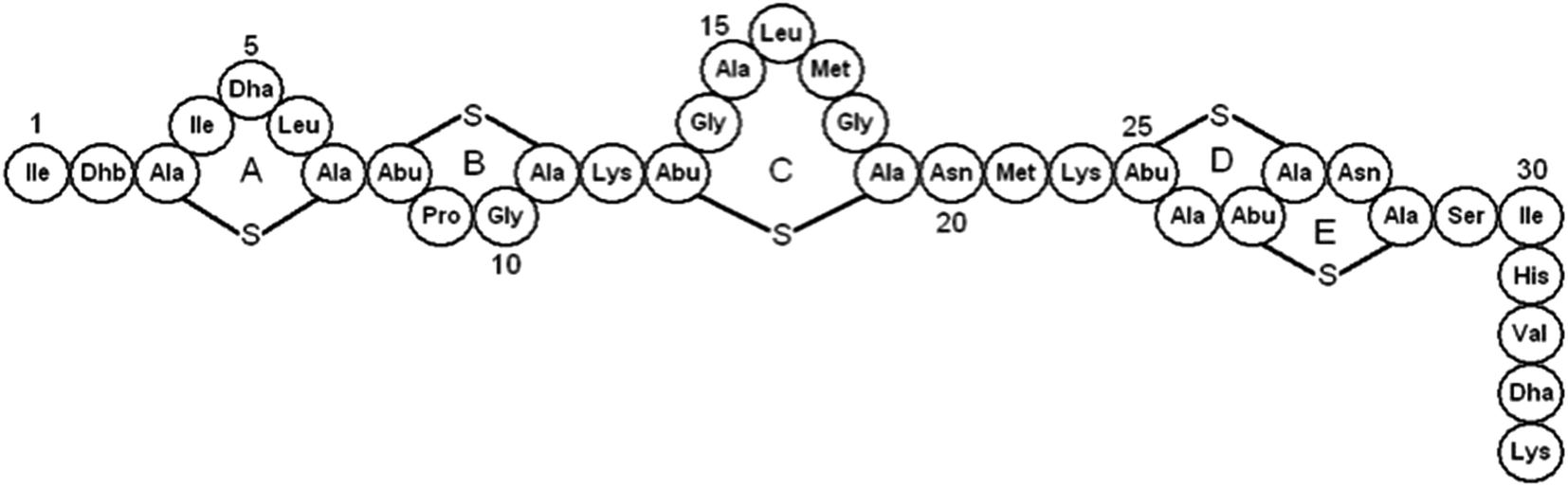 | ||
| Fig. 1 Schematic of nisin, showing the sequence and the location of the thioether rings. | ||
Functional membrane-lytic complexes form between nisin and its target lipid II with 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry in a pyrophosphate-mediated process and are proposed to assemble into higher order oligomeric structures.20 An NMR study of 1:1 nisin–lipid II model in DMSO indicates N-terminal engagement by rings A and B of nisin with the pyrophosphate group in lipid II.21 Structural information from the biologically relevant nisin–lipid II 2:1 membrane complexes is lacking at present.
:1 stoichiometry in a pyrophosphate-mediated process and are proposed to assemble into higher order oligomeric structures.20 An NMR study of 1:1 nisin–lipid II model in DMSO indicates N-terminal engagement by rings A and B of nisin with the pyrophosphate group in lipid II.21 Structural information from the biologically relevant nisin–lipid II 2:1 membrane complexes is lacking at present.
The CHARMM force field,22 which describes biological macromolecules such as proteins,22,23 nucleic acids24 and their ligands25 in aqueous or membrane environments26,27 is widely used in atomistic molecular dynamics (MD) simulations. Despite the research interest in lantibiotics and their long-term industrial use, simulation studies have been hindered by limited theoretical characterization of the dehydrated residues Dha and Dhb. Parameters have been developed for Dha28 for the AMBER force field29 but these have been used to study the propensity of dehydroamino residues to form β-turns in de novo peptides30,31 and not for the study of lantibiotics. More recently, Siodłak et al.32 calculated the complete Ramachandran plot for the dehydroalanine dipeptide at the B3LYP/6-31+G** level of theory, again with a focus on peptide design.
In this work we computationally characterize Dha and Dhb and report a new set of parameters for the CHARMM force field to describe Dha and Dhb in a polypeptide chain. To ensure consistency with the CHARMM36 force field, the parameterization procedure33 followed here is the same as that used for the protein, lipid and small-molecule parameter sets. The new parameters have enabled us to perform the first simulation of nisin interacting with a model lipid membrane of palmitoyloleoyl phosphatidylglycerol (POPG) and palmitoyloleoyl phosphatidylethanolamine (POPE). These simulations show that nisin readily binds to membrane surfaces through specific hydrogen bonding, in agreement with NMR data.12 The parameters and simulations presented here form a platform for further study by MD simulation of interactions between lipid II and nisin in order to predict the structure and formation mechanism of the nisin–lipid II membrane spanning pore.
Methods
Target molecule and initial guess
The parameters were produced following the established procedure for introducing new chemical groups to the CHARMM force field.22,25,33 The target molecule for the parameterization was the dehydroalanine dipeptide with an acetylated N-terminus and N-methylamide C-terminus (Fig. 2). These end groups were chosen over simple methyl groups so that the effect, if any, of the double bond to the α-carbon on the neighbouring peptide bonds would be accounted for. New atom types, with new charges and parameters, were created for atoms N8, H9, C6, C7, H7, H8, C5 and O9 and adjusted during the parameterization. The remaining atom types were kept the same as their analogies from the CHARMM protein set and were not adjusted. | ||
| Fig. 2 Target molecule for dehydroalanine with the atom labeling used in the parameterization. | ||
The initial stream file, containing the starting guess for the topology, charges and parameters, of the dehydroalanine dipeptide was produced using the ParamChem34 interface (https://www.paramchem.org). The ParamChem project aims to automate the parameterization of new molecular species in CHARMM by making analogies with pre-existing chemical groups and assigning a penalty that is a measure of the confidence of the analogy. Due to the unusual double bond between Cα and Cβ there were no suitable analogies present in the CHARMM force field, leading to large ParamChem penalties of 163 (parameter) and 170 (charge). Penalties under 10 are considered reasonable, confirming the need for further parameterization of the dehydrated residues.
Target data
The CHARMM potential energy function is given by
 | (1) |
The bond, angle and Urey–Bradley terms (1,3-pairs) are harmonic potentials each with associated force constants K, to describe bond stretching and bond angle bending. The dihedral potential describes the energy associated with bond twisting and improper dihedrals are artificial potentials that are used to restrict the conformation of a group consisting of a central atom bonded to three others. The UB and improper terms are rarely defined for new atom types but remain in the parameter set for older atom types or situations when the sum of the other terms cannot satisfactory describe a chemical group. The final term in the CHARMM force field, the CMAP term,23 is a correction applied to the φ and ψ angles of the protein backbone to redress systematic errors in secondary structure. As nisin does not form any secondary structure elements, this term (along with the UB term) was not considered in our parameterization of the new Dha and Dhb residues.
The force constants, Kb and Kθ, were optimized to reproduce the bond lengths and bond angles of an MP2/6-31G* optimized geometry of the dehydroalanine dipeptide. This level of quantum chemistry has been used consistently23,25,27 for the development of the CHARMM force field over recent years. The initial partial charges, q, were chosen by analogy with pre-existing chemical groups and optimized to reproduce the interaction distances and energies, calculated at the HF/6-31G* level, between hydrogen bond donors and acceptors and single water molecules (Fig. 3) in the TIP3P geometry.35 The HF/6-31G* interaction distances and energies were decreased by 0.2 Å and scaled by 1.16 respectively, to maintain consistency with the existing parameters in the CHARMM force field. Further details of the partial charge optimization procedure are available in the literature.25,33 For three of the donor–acceptor interactions (Dha N10H11 – OW, Dha O9 – HW and Dha O3 – HW, where OW is the oxygen of the water molecule and HW is a hydrogen) the dipeptide was in the lowest energy conformation, corresponding to (ϕ,φ) of (−172°, 154°). When in this conformation, the fourth donor–acceptor interaction (Dha N8H9 – OW) had an overlap where the test water molecule interacting with Dha N8H9 was also interacting with Dha O3. To resolve this issue, the interaction energy and distance for the partial charges were calculated when the dipeptide was in an alternative conformation corresponding to (−58°, 30°); this conformation was identified as the second lowest energy conformation by Siodłak et al.32 Dihedral parameters were fitted to reproduce relaxed scans at the MP2/6-31G*//MP2/cc-pVTZ level through a combination of the fitting program of Guvench and MacKerell36 and further optimization ‘by hand’ of C4–N8–C6–C5 and C4–N8–C6–C7 to ensure agreement in the low energy regions. The parameters for Dhb were developed by analogy with Dha, with the additional methyl group added by analogy with hydrocarbons in the CGenFF parameter set.25 All quantum chemistry calculations were performed using Q-Chem;37 all empirical force field minimization calculations were performed using CHARMM.38,39
 | ||
| Fig. 3 (Left) Interactions between water molecules and N10H11, O9 and O3 of the dehydroalanine dipeptide in the optimized geometry (water molecules superimposed onto the same image for illustration). (Right) Alternative conformation used to calculate the interaction between a single water molecule and N8H9. | ||
Chemical shift calculations
Chemical shifts of the dehydrated residues were calculated by density-functional theory (DFT) and compared to experimental values. The calculations were performed using the DALTON quantum chemistry program40,41 by determining the differences in absolute isotropic shielding constants between those calculated for each compound and the relevant reference systems: TMS (1H NMR), H2O (17O NMR) NH3 (14N NMR). All calculations were performed using the KT3 functional,42 which was specifically designed for the calculation of absolute shielding constants, along with the aug-pcS-2 basis sets of Jensen.43 The basis sets employed were specifically designed for use in DFT and have been optimized for the calculation of NMR shielding constants.43 For all calculations of chemical shifts we used the MP2/6-31G* optimized geometry, which was also used as the target data for the bond lengths and angles.Molecular dynamics simulations
Using CHARMM-GUI,44,45 a solvated lipid bilayer was generated with a composition of 3:1 POPG:POPE lipids, to represent Gram-positive bacterial membranes.46 Each leaflet of the membrane contained 37 POPE lipids and 111 POPG lipids and the area of the membrane surface was 95 × 95 Å in the x–y plane. This choice of system size is based upon similar MD studies of lipid II inserted in a bilayer46 and the interaction between vancomycin and lipid II.47 The depth of the water solvent layer above and below the bilayer was 30 Å and contained 222 neutralizing potassium ions. Three independent simulations were conducted. The starting coordinates were taken from models in the Protein Data Bank (PDB) entry 1WCO, the NMR structure of a single nisin molecule in complex with truncated lipid II. The lipid II molecule was deleted and the remaining nisin molecule orientated so that the principal geometric axis was parallel with the x-axis, the next largest direction was parallel with the y-axis and the atom with the minimal z-direction coordinate was 1 Å above the nearest lipid head group, in the water layer. Any water molecules or potassium ions that overlapped with the nisin molecule were deleted. The building of the molecular system and the post-production dynamics analysis were performed using CHARMM.38,39
All minimization, equilibration, and production dynamics were performed using NAMD48 with a time step of 2 fs and the CHARMM36 force field with the new parameters developed here. Periodic boundary conditions were applied using a cuboid cell with starting dimensions of 95 Å × 95 Å × 100 Å. The SHAKE algorithm was applied to all bonds to hydrogen atoms.49 The non-bonded cut-off was 12.0 Å and the non-bonded neighbour list was updated at every time step. Long-range electrostatics were treated using the particle-mesh Ewald method.50 Initial minimization was performed using the standard NAMD minimization algorithm, which is a combination of conjugate gradient and line search methods. The minimization was performed in stages with 300 steps with all atoms, except the lipid and lipid II tails, fixed in position; 300 steps with the nisin–lipid II complex fixed; 300 steps with the nisin molecule fixed and finally 300 steps where all atoms are able to relax in position. This was followed by a further 1000 steps of minimization and the first 0.5 ns phase of equilibration where the lipid tails are relaxed and all other atoms are fixed in position. In the second equilibration phase 2500 steps of minimization and a further 0.5 ns of equilibration was performed with the nisin molecule fixed. In the next equilibration stage all the atoms were relaxed for 2500 steps of minimization followed by 5 ns of heating and equilibration to 300 K in the NPT ensemble. Production dynamics were run for 100 ns at 300 K in the NPAT ensemble, where the surface area in the x–y plane was also kept constant to maintain a biologically relevant lipid density.27
A 100 ns simulation of nisin solvated in water was performed to compare with the conformational preferences of nisin interacting with a membrane. The starting coordinates were taken from models in the Protein Data Bank (PDB) entry 1WCO, with the lipid II molecule deleted. The nisin molecule was inserted into a water cube of 90 Å × 90 Å × 90 Å and containing four neutralizing chloride ions. The time step was 2 fs and the simulation used the CHARMM36 force field with the new parameters developed here. Periodic boundary conditions were applied using a cuboid cell. Other simulation details were as given in the preceding paragraph, except as noted below. Minimization was performed in two stages, with 10000 steps holding the peptide backbone fixed and 10000 steps where all atoms are able to relax in position. The system was heated to 300 K and equilibrated in the NPT ensemble for 10 ns. Production dynamics were run for 100 ns at 300 K.
Results
QM calculations on dehydroalanine dipeptide
The molecular orbitals from the MP2/6-31G*//MP2/cc-pVTZ geometry optimization and energy evaluation, characterize the electronic structure of the dehydroalanine dipeptide. The converged total energies for the two levels of theory are −491.672404651716 and −493.152241462216 Eh, respectively. The identification of the π molecular orbitals was aided by reference to a model five atom system calculated using the Simple Hückel Molecular Orbital Theory Calculator (http://www.chem.ucalgary.ca/SHMO/).51 Each carbon atom, and the oxygen, contributes one out-of-plane 2p electron and the nitrogen atom contributes two electrons. Thus, there are six π electrons and five π molecular orbitals. These were readily identified from the ab initio calculation (Fig. 4), with the highest occupied molecular orbital (HOMO) corresponding to the third π molecular orbital (approximately non-bonding, with two out-of-plane nodes). This conjugation system that includes the double bond between Cα and Cβ is highly unusual in naturally occurring peptides and has a strong influence on the chemical shifts calculated in the next subsection. | ||
| Fig. 4 The π molecular orbital diagram (left) with MP2 calculated orbital energies (in eV) and corresponding Hartree–Fock orbitals calculated for the model system. The Dha dipeptide (top right), and the calculated isovalue surface (corresponding to the points in space where the density takes a value of 0.02) for the third π molecular orbital of Dha (bottom right). | ||
Chemical shifts
Experimentally verifiable parameters that are dependent on the electron density distribution are the NMR chemical shift. Mean experimental values for the chemical shifts of Dha and Dhb were retrieved from the Biological Magnetic Resonance Data Bank (BMRB).52 Table 1 compares these values to the chemical shifts calculated by DFT, those predicted by the ADC labs NMR predictor module (http://www.adclabs.com) and those measured by Chan et al.53 in a study of the lantibiotic subtilin.| Atom | BMRB | Subtilin53 | Calculated | Predicted | |||||
|---|---|---|---|---|---|---|---|---|---|
| Dha | N | 121.9 | — | 109.4 | — | ||||
| Cα | 134.0 | — | 136.5 | 135.9 | |||||
| Cβ | 101.2 | — | 95.7 | 103.2 | |||||
| C | 167.3 | — | 164.6 | 170.4 | |||||
| HN | 9.21 | 9.69 | 8.73 | 8.92 | |||||
| Hβ | 5.58 | 5.97 | 5.65 | 5.74 | 5.04 | 5.96 | 5.93 | 6.02 | |
| Dhb | N | 127.5 | — | 109.4 | — | ||||
| Cα | 130.3 | — | 133.2 | 130.2 | |||||
| Cβ | 137.9 | — | 121.9 | 121.6 | |||||
| C | 168.9 | — | 166.1 | 170.5 | |||||
| HN | 10.5 | 8.74 | 8.27 | 8.28 | |||||
| Hβ | — | 6.91 | 6.12 | 5.83 | |||||
Due to the double bond in the dehydrated residues, the chemical shifts of Cα and Cβ are very different from the values observed in the standard amino acid residues, which are usually between 45 to 65 ppm and 20 to 70 ppm, respectively. However, there are also differences in the shifts that characterize the peptide backbone. The amino protons show an upfield shift to almost 9.5 ppm from the usual values of between 7.5 and 8.5 ppm and the carbonyl 13C chemical shifts are below 170 ppm in contrast to the carbonyl shift values observed in the common amino acid residues in the region between 174 and 178 ppm.
CHARMM parameterization
Tables 2 and 3 compare bond lengths and angles from MP2/6-31G* geometry optimization with those calculated from an energy minimization using the new parameters. It is recommended25 that the bond lengths from the CHARMM optimization are within 0.03 Å of the QM optimized value and the valence angles within 3°. Both show close agreement between the CHARMM and QM values; the average difference in bond length in Table 2 is 0.02 ± 0.01 Å and the average difference in bond angle in Table 3 is 1 ± 1°. Table 4 reports the result of the partial charge optimization. The recommended25 minimum difference between the QM values and the optimized CHARMM parameters is 0.2 kcal mol−1 for the interaction energies and 0.1 Å for the interaction distance. Although the differences in interaction distance for Dha N10H11 and interaction energy for Dha N8H9 slightly exceed this, similar disagreement has been acceptable in major parameterization efforts such as CGenFF.25 Fig. 5 is an example of a dihedral scan showing the MP2/6-31G* potential energy surface with respect to this coordinate and the surface calculated using the initial guess and the new CHARMM parameters. The disagreement in the low energy region, about ±180°, between the initial guess and the optimized parameters and the QM surface illustrates the necessity of applying the full parameterization procedure to the Dha and Dhb residues.| Bond | MP2/6-31G* geometry optimisation (Å) | Optimised CHARMM parameters (Å) | Difference (Å) |
|---|---|---|---|
| C6–C7 | 1.32 | 1.35 | 0.03 |
| C6–N8 | 1.40 | 1.41 | 0.01 |
| C5–C6 | 1.51 | 1.53 | 0.02 |
| C5–O9 | 1.21 | 1.23 | 0.03 |
| C4–N8 | 1.36 | 1.34 | 0.02 |
| O3–C4 | 1.20 | 1.22 | 0.03 |
| C2–C4 | 1.51 | 1.48 | 0.04 |
| C5–N10 | 1.35 | 1.36 | 0.01 |
| C1–N10 | 1.45 | 1.45 | 0.00 |
| C1–H1 | 1.08 | 1.11 | 0.03 |
| C1–H2 | 1.08 | 1.11 | 0.03 |
| C1–H3 | 1.09 | 1.11 | 0.03 |
| C2–H4 | 1.09 | 1.11 | 0.03 |
| C2–H5 | 1.09 | 1.11 | 0.02 |
| C2–H6 | 1.08 | 1.11 | 0.03 |
| C7–H7 | 1.07 | 1.10 | 0.03 |
| C7–H8 | 1.07 | 1.10 | 0.03 |
| N8–H9 | 1.00 | 1.00 | 0.00 |
| N10–H11 | 0.99 | 0.99 | 0.00 |
| Angle | MP2/6-31G* geometry optimisation (°) | Optimised CHARMM parameters (°) | Difference (°) |
|---|---|---|---|
| H4–C2–H5 | 109 | 109 | 0 |
| H4–C2–H6 | 109 | 109 | 0 |
| H6–C2–H4 | 108 | 109 | 1 |
| H4–C2–C4 | 110 | 110 | 0 |
| H5–C2–C4 | 109 | 110 | 1 |
| H6–C2–C4 | 112 | 110 | 2 |
| C2–C4–O3 | 122 | 121 | 2 |
| C2–C4–N8 | 114 | 115 | 1 |
| C4–N8–H9 | 119 | 119 | 0 |
| C4–N8–C6 | 128 | 130 | 2 |
| N8–C6–C7 | 127 | 128 | 1 |
| N8–C6–C5 | 110 | 110 | 0 |
| C6–C7–H7 | 122 | 123 | 1 |
| C6–C7–H8 | 120 | 119 | 1 |
| H7–C7–H8 | 118 | 118 | 0 |
| C6–C5–O9 | 120 | 122 | 2 |
| C6–C5–N10 | 118 | 119 | 2 |
| C5–N10–H11 | 119 | 120 | 2 |
| C5–N10–C1 | 121 | 122 | 1 |
| N10–C1–H1 | 112 | 111 | 1 |
| N10–C1–H2 | 109 | 111 | 2 |
| N10–C1–H3 | 110 | 111 | 1 |
| H1–C1–H2 | 109 | 108 | 1 |
| H1–C1–H3 | 109 | 108 | 1 |
| H2–C1–H3 | 108 | 108 | 1 |
| Interaction | Adjusted interaction distance (Å) | Adjusted interaction energy (kcal mol−1) | Optimised CHARMM parameters | Distance difference (Å) | Energy difference (kcal mol−1) | |
|---|---|---|---|---|---|---|
| Distance (Å) | Energy (kcal mol−1) | |||||
| Dha N10H11 – OW | 1.97 | −6.59 | 2.24 | −6.52 | 0.27 | 0.07 |
| Dha O9 – HW | 1.83 | −7.11 | 1.73 | −7.11 | 0.10 | 0.00 |
| Dha N8H9 – OW | 1.92 | −7.39 | 1.95 | −7.04 | 0.03 | 0.35 |
| Dha O3 – HW | 1.83 | −6.20 | 1.78 | −6.20 | 0.05 | 0.00 |
 | ||
| Fig. 5 An example dihedral scan around the N8–C6–C5–N10 (ϕ dihedral angle) showing the MP2/6-31G*//MP2/cc-pVTZ surface (red solid line with pluses), the CHARMM initial guess (blue dotted line with asterisks) and the CHARMM surface after the parameters have been optimized (green dashed line with crosses). | ||
MD simulation
The thermodynamic stability of the MD simulations was confirmed by examining the kinetic energy, potential energy, total energy, temperature and pressure (data not shown). Nisin does not have any secondary structure elements such as α-helices or β-sheets, but there is some structure imposed by the steric restrictions of the thioether rings. Table 5 shows the (ϕ,φ) angles of the residues that form the interior of the rings during the membrane simulation. In agreement with NMR structure of nisin in micelles,18 rings B and D form type-II β-turns and ring E forms a distorted type-I β-turn, using the definitions of Hutchinson and Thornton.54 This contrasts with the simulation of nisin in water (Table 6), where ring B adopts a type-II β-turn but the other rings have no recognised regular structure, indicating that nisin in water has more conformational freedom and that the interaction with the membrane constrains the rings.| Residue number | ϕ (°) | Std Dev (°) | φ (°) | Std Dev (°) | |
|---|---|---|---|---|---|
| Ring A | 4 | −101 | 39 | 8 | 38 |
| 5 | −176 | 15 | −124 | 14 | |
| 6 | −82 | 16 | 102 | 31 | |
| Ring B | 9 | −80 | 15 | 85 | 23 |
| 10 | 76 | 17 | 34 | 21 | |
| Ring C | 14 | −62 | 13 | 155 | 17 |
| 15 | −73 | 14 | −35 | 13 | |
| 16 | −63 | 9 | −43 | 8 | |
| 17 | −98 | 11 | −2 | 11 | |
| 18 | 63 | 11 | 25 | 11 | |
| Ring D | 24 | −68 | 10 | 100 | 10 |
| 25 | 81 | 16 | 24 | 15 | |
| Ring E | 26 | −96 | 15 | 1 | 68 |
| 27 | −162 | 81 | 19 | 28 |
| Residue number | ϕ (°) | Std Dev (°) | φ (°) | Std Dev (°) | |
|---|---|---|---|---|---|
| Ring A | 4 | −82 | 21 | −24 | 21 |
| 5 | −178 | 10 | 155 | 10 | |
| 6 | 40 | 51 | 62 | 32 | |
| Ring B | 9 | −67 | 12 | 86 | 62 |
| 10 | 106 | 68 | 19 | 21 | |
| Ring C | 14 | −99 | 59 | 168 | 21 |
| 15 | −55 | 34 | −43 | 32 | |
| 16 | −83 | 21 | −11 | 27 | |
| 17 | −95 | 16 | −5 | 17 | |
| 18 | 81 | 19 | 27 | 20 | |
| Ring D | 24 | −112 | 20 | 109 | 15 |
| 25 | 81 | 14 | 36 | 12 | |
| Ring E | 26 | −92 | 16 | 108 | 14 |
| 27 | 63 | 11 | 40 | 15 |
Receptor recognition by nisin involves the formation of multiple hydrogen bonds55 and hydrogen bonding is likely to be an important component of membrane interactions. To investigate this, a hydrogen bonding analysis was performed between nisin and the lipid head groups. The cutoff for the hydrogen-acceptor distance was 2.8 Å, there was no cutoff for the bond angle, and structures were sampled every 4 ps. Table 7 shows the percentage of trajectory frames where a hydrogen bond is formed between a polar hydrogen atom in nisin and oxygen atoms in the phosphate group of the lipid head groups. Interactions between nisin carbonyls and lipid head group hydroxyls were observed, but across the three trajectory runs the mean occupancy was 3% ± 4% and the mean lifetime was 12 ps ± 8 ps, indicating that they were not sustained interactions. The nisin N-terminus and Lys12 side chain perform a random walk across the membrane surface by forming hydrogen bonds with several lipid head groups. During trajectory 1 the N-terminus group makes a hydrogen bond with five different lipids, one POPE and four POPG; during trajectory 2 there is hydrogen bonding to five POPG lipids; and during trajectory 3 hydrogen bonds are formed with eight lipids, one POPE and seven POPG. Similarly, the side chain of Lys12 forms hydrogen bonds with eleven, seven and five different lipids during trajectories 1, 2 and 3, respectively.
| Atom | Trajectory | ||
|---|---|---|---|
| 1 | 2 | 3 | |
| Occupancy | |||
| Ile1 HT1 | 40% | 47% | 36% |
| Ile1 HT2 | 45% | 30% | 44% |
| Ile1 HT3 | 44% | 27% | 38% |
| Dhb2 HN | 24% | 25% | 27% |
| Dha5 HN | 0% | 16% | 0% |
| Leu6 HN | 0% | 18% | 0% |
| Cys7 HN | 0% | 33% | 0% |
| Abu8 HN | 0% | 23% | 14% |
| Lys12 HN | 0% | 59% | 15% |
| Lys12 HZ1 | 32% | 49% | 39% |
| Lys12 HZ2 | 38% | 56% | 32% |
| Lys12 HZ3 | 35% | 52% | 39% |
| Abu13 HN | 0% | 71% | 0% |
| Gly14 HN | 0% | 61% | 0% |
| Lys22 HN | 0% | 0% | 37% |
| Lys22 HZ1 | 0% | 26% | 40% |
| Lys22 HZ2 | 0% | 27% | 34% |
| Lys22 HZ3 | 0% | 26% | 37% |
| Abu23 HN | 0% | 27% | 3% |
| Ala24 HN | 0% | 19% | 0% |
| His31 HE2 | 10% | 0% | 18% |
| Lys34 HZ1 | 20% | 2% | 11% |
| Lys34 HZ2 | 22% | 1% | 10% |
| Lys34 HZ3 | 24% | 1% | 12% |
During the simulation of nisin in water an α-helical turn forms at the C-terminus, stabilized by a hydrogen bond between the carbonyl oxygen of Ser29 and the amide of Dha33. This hydrogen bond and structure is not observed for the simulation of nisin in the lipid environment.
To investigate the conformational preferences of the nisin in the aqueous and membrane environments, a clustering analysis of the (ϕ,φ) angles was performed using the algorithm described by Karpen et al.56 In this implementation the membership of each cluster is unique and all conformations in a cluster are within a 30° radius of each other. For the peptide interacting with the membrane 57 clusters were produced, of which the most populated contained 20% of the trajectory frames (Fig. 6). When the peptide was in water there were 112 clusters and the most populated contained just 6% of the trajectory frames, illustrating the greater conformational diversity of nisin in water due to the conformational freedom in the z-direction and the absence of the constraining hydrogen bonds between the lysine tails and the membrane.
 | ||
| Fig. 6 (Left) Trajectory snapshot from the most populated cluster showing the periodic cell (water omitted for clarity); (right) the same trajectory snapshot showing hydrogen bonding between the nisin N-terminus, Lys12 and oxygen atoms from the phosphate moiety of lipid head groups, phosphate atoms of lipid head groups included to illustrate membrane surface. | ||
Discussion
The presence of an unsaturated carbon at the α-position in Dha and Dhb leads to an unusual redistribution of electron density across neighbouring peptide bonds. The experimental, calculated and predicted chemical shifts of Cα and Cβ of the dehydrated residues are 125–135 ppm and 88–138 ppm, respectively, compared with 45–65 ppm and 20–70 ppm in the standard amino residues. Delocalization of electron density from the nitrogen site towards the α-carbon is associated with an increase in electron density in the carbonyl region. This is in agreement with experimentally observed deshielding at the amino protons, which gives rise to a significant upfield shift in Dha and Dhb.16,57,58 Concomitantly, carbonyl 13C chemical shifts from Dha and Dhb are below 170 ppm in contrast to carbonyl shift values observed from the common amino acid residues in the region between 174 and 178 ppm. This electronic delocalization is reflected in the partial charges of the new parameters of dehydroamino residues. For standard amino residues in the CHARMM force field, the charges of the grouping consisting of the backbone atoms N, HN, Cα and Hα are −0.47, 0.31, 0.07 and 0.09, respectively; for the equivalent grouping from the dehydroamino parameters the partial charges of N, HN and Cα are −0.67, 0.36 and 0.31. Similarly, the partial charges of the backbone carbonyl group are −0.51 for oxygen and 0.51 for carbon for standard amino residues in the CHARMM force field and ±0.635 for the dehydroamino residues.Nisin binding to lipid membranes
The new parameters were used in an all-atom MD simulation of nisin in the presence of a hydrated model membrane of POPE/POPG, which revealed association of the peptide with the membrane surface. N-terminal association of the peptide with POPE/POPG bilayer revealed hydrogen bonding with oxygen atoms of the phosphate moiety of individual lipid head groups. During the simulations, the N-terminus hydrogen-bonded to both lipid species and the ε-amine of Lys12 formed hydrogen bonds with a similar preference for POPG. This supports a model in which receptor recognition is preceded by engagement of membrane lipid head groups by cationic nisin residues 1 and 12, followed by a random walk across (Fig. 6) the membrane surface towards triply negatively charged lipid II molecules. Several experimental observations are consistent with this model, such as decreased activity associated with N-terminal succinylation59 and methylation,5 where the N-terminus is no longer available as hydrogen bond donor. Similarly, extension of the nisin N-terminus has a detrimental effect on biological activity,60,61 where the presence of the leaders stops the amino group of Ile1 from interacting with the membrane, as it would have fewer hydrogen bond donors to mediate the interaction and there would be steric limitations due to the peptide backbone. Compared with native nisin, a Lys12Leu variant associates less strongly with the surface of a model membrane system that did not contain lipid II.62 This non-conservative mutation would prevent the formation of hydrogen bonds between the side chain of residue 12 and the membrane and would explain the reduced interaction.Conclusions
We present a set of CHARMM force field parameters for dehydroalanine and dehydrobutyrine, commonly present in lanthionine antibiotics. These were developed following the established protocol for adding new chemical moieties to the CHARMM force field and are compatible with the parameters for biological macromolecules such as proteins, lipids, nucleic acids and small drug-like molecules. The new parameters were verified in MD studies of the nisin molecule in lipid membrane and aqueous environments. The results showed good agreement with experimental and theoretically predicted NMR data, as well as with the results of previous studies of nisin mutants. The findings support the idea that nisin associates with a bacterial membrane in the absence of lipid II, via hydrogen bonding with lysine side chains, and performs a random walk across the membrane surface. We are currently using these parameters to investigate the interaction between native nisin and lipid II in a lipid bilayer.Acknowledgements
We are grateful for access to the University of Nottingham High Performance Computing (HPC) Facility and the MidPlus Regional Centre of Excellence for Computational Science, Engineering and Mathematics under EPSRC grant EP/K000128/1. Whilst undertaking this work ERT was supported by EPSRC Doctoral Prize funding. SM is supported by a University of Nottingham interdisciplinary HPC PhD studentship. AMT is grateful for support from the Royal Society University Research Fellowship scheme.References
- J. M. Willey and W. A. van der Donk, Lantibiotics: Peptides of diverse structure and function, in Annual Review of Microbiology, Annual Reviews, Palo Alto, 2007, pp. 477–501 Search PubMed.
- S. M. Asaduzzaman and K. Sonomoto, Lantibiotics: Diverse activities and unique modes of action, J. Biosci. Bioeng., 2009, 107, 475–487 CrossRef CAS PubMed.
- G. Bierbaum and H. G. Sahl, Lantibiotics: Mode of Action, Biosynthesis and Bioengineering, Curr. Pharm. Biotechnol., 2009, 10, 2–18 CAS.
- J. Dischinger, I. Wiedemann, G. Bierbaum and H.-G. Sahl, Chapter 19-Lantibiotics, in Handbook of Biologically Active Peptides, ed. A. J. Kastin, Academic Press, Boston, 2nd edn, 2013, pp. 119–128 Search PubMed.
- B. B. Bonev, E. Breukink, E. Swiezewska, B. De Kruijff and A. Watts, Targeting extracellular pyrophosphates underpins the high selectivity of nisin, FASEB J., 2004, 18, 1862–1869 CrossRef CAS PubMed.
- E. Breukink, I. Wiedemann, C. van Kraaij, O. P. Kuipers, H. G. Sahl and B. de Kruijff, Use of the cell wall precursor lipid II by a pore-forming peptide antibiotic, Science, 1999, 286, 2361–2364 CrossRef CAS.
- I. Wiedemann, E. Breukink, C. van Kraaij, O. P. Kuipers, G. Bierbaum, B. de Kruijff and H. A. Sahl, Specific binding of nisin to the peptidoglycan precursor lipid II combines pore formation and inhibition of cell wall biosynthesis for potent antibiotic activity, J. Biol. Chem., 2001, 276, 1772–1779 CrossRef CAS PubMed.
- A. J. Hyde, J. Parisot, A. McNichol and B. B. Bonev, Nisin-induced changes in Bacillus morphology suggest a paradigm of antibiotic action, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19896–19901 CrossRef CAS PubMed.
- W. C. Chan, H. M. Dodd, N. Horn, K. Maclean, L. Y. Lian, B. W. Bycroft, M. J. Gasson and G. C. K. Roberts, Structure–activity relationships in the peptide antibiotic nisin: Role of dehydroalanine 5, Appl. Environ. Microbiol., 1996, 62, 2966–2969 CAS.
- I. M. Gut, S. R. Blanke and W. A. van der Donk, Mechanism of Inhibition of Bacillus anthracis Spore Outgrowth by the Lantibiotic Nisin, ACS Chem. Biol., 2011, 6, 744–752 CrossRef CAS PubMed.
- E. Breukink, C. vanKraaij, R. A. Demel, R. J. Siezen, O. P. Kuipers and B. deKruijff, The C-terminal region of nisin is responsible for the initial interaction of nisin with the target membrane, Biochemistry, 1997, 36, 6968–6976 CrossRef CAS PubMed.
- B. B. Bonev, W. C. Chan, B. W. Bycroft, G. C. K. Roberts and A. Watts, Interaction of the lantibiotic nisin with mixed lipid bilayers: A P-31 and H-2 NMR study, Biochemistry, 2000, 39, 11425–11433 CrossRef CAS PubMed.
- W. C. Chan, L. Y. Lian, B. W. Bycroft and G. C. K. Roberts, Confirmation of the structure of nisin by complete H-1-NMR resonance assignment in aqueous and dimethyl sulfoxide solution, J. Chem. Soc., Perkin Trans. 1, 1989, 2359–2367 RSC.
- M. Slijper, C. W. Hilbers, R. N. H. Konings and F. J. M. Vandeven, NMR studies of lantibiotics: assignment of the H-1-NMR spectrum of nisin and identification of interresidual contacts, FEBS Lett., 1989, 252, 22–28 CrossRef CAS.
- F. J. M. van de Ven, H. W. van den Hooven, R. N. H. Konings and C. W. Hilbers, NMR-studies of lantibiotics – the structure of nisin in aqueous solution, Eur. J. Biochem., 1991, 202, 1181–1188 CrossRef CAS PubMed.
- L. Y. Lian, W. C. Chan, S. D. Morley, G. C. K. Roberts, B. W. Bycroft and D. Jackson, Solution structures of nisin-A and its 2 major degradation products determined by NMR, Biochem. J., 1992, 283, 413–420 CAS.
- H. W. van den Hooven, F. Fogolari, H. S. Rollema, R. N. H. Konings, C. W. Hilbers and F. J. M. van de Ven, NMR and circular dichroism studies of the lantibiotic nisin in nonaqueous environments, FEBS Lett., 1993, 319, 189–194 CrossRef CAS.
- H. W. Van Den Hooven, C. C. M. Doeland, M. Van De Kamp, R. N. H. Konings, C. W. Hilbers and F. J. M. Van De Ven, Three-dimensional structure of the lantibiotic nisin in the presence of membrane-mimetic micelles of dodecylphosphocholine and of sodium dodecylsulphate, Eur. J. Biochem., 1996, 235, 382–393 CAS.
- E. R. Turpin, B. B. Bonev and J. D. Hirst, Stereoselective Disulfide Formation Stabilizes the Local Peptide Conformation in Nisin Mimics, Biochemistry, 2010, 49, 9594–9603 CrossRef CAS PubMed.
- H. E. Hasper, B. de Kruijff and E. Breukink, Assembly and stability of nisin–lipid II pores, Biochemistry, 2004, 43, 11567–11575 CrossRef CAS PubMed.
- H.-J. Hsu, H.-J. Chang, H.-P. Peng, S.-S. Huang, M.-Y. Lin and A.-S. Yang, Assessing computational amino acid β-turn propensities with a phage-displayed combinatorial libray and directed evolution, Structure, 2006, 14, 1499–1510 CrossRef CAS PubMed.
- A. D. MacKerell Jr, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, All-atom empirical potential for molecular modeling and dynamics studies of proteins, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef PubMed.
- A. D. Mackerell Jr, M. Feig and C. L. Brooks III, Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations, J. Comput. Chem., 2004, 25, 1400–1415 CrossRef PubMed.
- N. Foloppe and J. A. D. MacKerell, All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data, J. Comput. Chem., 2000, 21, 86–104 CrossRef CAS.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. M. Mackerell Jr, CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields, J. Comput. Chem., 2009, 31, 671–690 Search PubMed.
- S. E. Feller and A. D. MacKerell Jr, An improved empirical potential energy function for molecular simulations of phospholipids, J. Phys. Chem. B, 2000, 104, 7510–7515 CrossRef CAS.
- J. B. Klauda, R. M. Venable, J. A. Freites, J. W. O'Connor, D. J. Tobias, C. Mondragon-Ramirez, I. Vorobyov, A. D. MacKerell Jr and R. W. Pastor, Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types, J. Phys. Chem. B, 2010, 114, 7830–7843 CrossRef CAS PubMed.
- G. Alagona and C. Ghio, Force fields parameters for molecular mechanical simulation of dehydroamino acid residues, J. Comput. Chem., 1991, 12, 934–942 CrossRef CAS.
- D. A. Case, T. E. Cheatham III, T. Darden, H. Gohlke, R. Luo, K. M. Merz Jr, A. Onufriev, C. Simmerling, B. Wang and R. Woods, The Amber biomolecular simulation programs, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- C. Aleman and J. J. Perez, A conformational study of the dehydroalanine: dipeptides and homopolypeptides, Biopolymers, 1993, 33, 1811–1817 CrossRef CAS PubMed.
- P. Mathur, S. Ramakumar and V. S. Chauhan, Peptide design using alpha,beta-dehydro amino acids: From beta-turns to helical hairpins, Biopolymers, 2004, 76, 150–161 CrossRef CAS PubMed.
- D. Siodłak, M. A. Broda and B. Rzeszotarska, Conformational analysis of α,β-dehydropeptide models at the HF and DFT levels, J. Mol. Struct.: THEOCHEM, 2004, 668, 75–85 CrossRef PubMed.
- A. D. Mackerell Jr, Atomistic Models and Force Fields, in Computational Biochemistry and Biophysics, ed. O. M. Becker, A. D. Mackerell Jr, B. Roux, and M. Watanabe, Marcel Dekker, Inc., New York, 2001 Search PubMed.
- K. Vanommeslaeghe and A. D. MacKerell, Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing, J. Chem. Inf. Model., 2012, 52, 3144–3154 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79, 926–936 CrossRef CAS PubMed.
- O. Guvench and A. D. MacKerell Jr, Automated conformational energy fitting for force-field development, J. Mol. Model., 2008, 18, 667–679 CrossRef PubMed.
- Y. Shao, L. F. Molnar, Y. Jung, J. Kussmann, C. Ochsenfeld, S. T. Brown, A. T. B. Gilbert, L. V. Slipchenko, S. V. Levchenko, D. P. O'Neill, R. A. DiStasio Jr, R. C. Lochan, T. Wang, G. J. O. Beran, N. A. Besley, J. M. Herbert, C. Yeh Lin, T. Van Voorhis, S. Hung Chien, A. Sodt, R. P. Steele, V. A. Rassolov, P. E. Maslen, P. P. Korambath, R. D. Adamson, B. Austin, J. Baker, E. F. C. Byrd, H. Dachsel, R. J. Doerksen, A. Dreuw, B. D. Dunietz, A. D. Dutoi, T. R. Furlani, S. R. Gwaltney, A. Heyden, S. Hirata, C.-P. Hsu, G. Kedziora, R. Z. Khalliulin, P. Klunzinger, A. M. Lee, M. S. Lee, W. Liang, I. Lotan, N. Nair, B. Peters, E. I. Proynov, P. A. Pieniazek, Y. Min Rhee, J. Ritchie, E. Rosta, C. David Sherrill, A. C. Simmonett, J. E. Subotnik, H. Lee Woodcock Iii, W. Zhang, A. T. Bell, A. K. Chakraborty, D. M. Chipman, F. J. Keil, A. Warshel, W. J. Hehre, H. F. Schaefer III, J. Kong, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Advances in methods and algorithms in a modern quantum chemistry program package, Phys. Chem. Chem. Phys., 2006, 8, 3172–3191 RSC.
- B. R. Brooks, R. E. Bruccoleri, D. J. Olafson, D. J. States, S. Swaminathan and M. Karplus, CHARMM: A program for macromolecular energy, minimization, and dynamics calculations, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- B. R. Brooks, C. L. Brooks, A. D. Mackerell Jr, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, CHARMM: The Biomolecular Simulation Program, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS PubMed.
- DALTON, a molecular electronic structure program, Release DALTON2013.3 (2013), http://daltonprogram.org.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani, P. Dahle, E. K. Dalskov, U. Ekström, T. Enevoldsen, J. J. Eriksen, P. Ettenhuber, B. Fernández, L. Ferrighi, H. Fliegl, L. Frediani, K. Hald, A. Halkier, C. Hättig, H. Heiberg, T. Helgaker, A. C. Hennum, H. Hettema, E. Hjertenæs, S. Høst, I.-M. Høyvik, M. F. Iozzi, B. Jansík, H. J. A. Jensen, D. Jonsson, P. Jørgensen, J. Kauczor, S. Kirpekar, T. Kjærgaard, W. Klopper, S. Knecht, R. Kobayashi, H. Koch, J. Kongsted, A. Krapp, K. Kristensen, A. Ligabue, O. B. Lutnæs, J. I. Melo, K. V. Mikkelsen, R. H. Myhre, C. Neiss, C. B. Nielsen, P. Norman, J. Olsen, J. M. H. Olsen, A. Osted, M. J. Packer, F. Pawlowski, T. B. Pedersen, P. F. Provasi, S. Reine, Z. Rinkevicius, T. A. Ruden, K. Ruud, V. V. Rybkin, P. Sałek, C. C. M. Samson, A. S. de Merás, T. Saue, S. P. A. Sauer, B. Schimmelpfennig, K. Sneskov, A. H. Steindal, K. O. Sylvester-Hvid, P. R. Taylor, A. M. Teale, E. I. Tellgren, D. P. Tew, A. J. Thorvaldsen, L. Thøgersen, O. Vahtras, M. A. Watson, D. J. D. Wilson, M. Ziolkowski and H. Ågren, The Dalton quantum chemistry program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 269–284 CrossRef CAS.
- T. W. Keal and D. J. Tozer, A semiempirical generalized gradient approximation exchange-correlation functional, J. Chem. Phys., 2004, 121, 5654–5660 CrossRef CAS PubMed.
- F. Jensen, Basis Set Convergence of Nuclear Magnetic Shielding Constants Calculated by Density Functional Methods, J. Chem. Theory Comput., 2008, 4, 719–727 CrossRef CAS.
- S. Jo, T. Kim and W. Im, Automated Builder and Database of Protein/Membrane Complexes for Molecular Dynamics Simulations, PLoS One, 2007, 2, e880 Search PubMed.
- S. Jo, J. B. Lim, J. B. Klauda and W. Im, CHARMM-GUI Membrane Builder for Mixed Bilayers and Its Application to Yeast Membranes, Biophys. J., 2009, 97, 50–58 CrossRef CAS PubMed.
- A. Chugunov, D. Pyrkova, D. Nolde, A. Polyansky, V. Pentkovsky and R. Efremov, Lipid-II forms potential “landing terrain” for lantibiotics in simulated bacterial membrane, Sci. Rep., 2013, 3 DOI:10.1038/srep01678.
- Z. Jia, M. L. O'Mara, J. Zuegg, M. A. Cooper and A. E. Mark, The Effect of Environment on the Recognition and Binding of Vancomycin to Native and Resistant Forms of Lipid II, Biophys. J., 2011, 101, 2684–2692 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, Scalable Molecular Dynamics with NAMD, J. Comput. Chem, 2005, 26, 1781–1802 CrossRef CAS PubMed.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes, J. Comp. Physiol., 1977, 23, 327–341 CAS.
- T. A. Darden, D. York and L. Pedersen, Particle-mesh Ewald: An Nlog(N) method for Ewald sums in large systems., J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS PubMed.
- A. Rauk, The Orbital Interaction Theory of Organic Chemistry, Wiley-Interscience, New York, 2nd edn, 2001 Search PubMed.
- E. L. Ulrich, H. Akutsu, J. F. Doreleijers, Y. Harano, Y. E. Ioannidis, J. Lin, M. Livny, S. Mading, D. Maziuk, Z. Miller, E. Nakatani, C. F. Schulte, D. E. Tolmie, R. Kent Wenger, H. Yao and J. L. Markley, BioMagResBank, Nucleic Acids Res., 2008, 36, D402–D408 CrossRef CAS PubMed.
- W. C. Chan, B. W. Bycroft, M. L. Leyland, L.-Y. Lian, J. C. Yang and G. C. K. Roberts, Sequence-specific resonance assignment and conformational analysis of subtilin by 2D NMR, FEBS Lett., 1992, 300, 56–62 CrossRef CAS.
- E. G. Hutchinson and J. M. Thornton, PROMOTIF – A program to identify and analyze structural motifs in proteins, Protein Sci., 1995, 5, 200–212 Search PubMed.
- S. T. D. Hsu, E. Breukink, E. Tischenko, M. A. G. Lutters, B. de Kruijff, R. Kaptein, A. Bonvin and N. A. J. van Nuland, The nisin–lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics, Nat. Struct. Mol. Biol., 2004, 11, 963–967 CAS.
- M. E. Karpen, D. J. Tobias and C. L. Brooks, Statistical clustering techniques for the analysis of long molecular dynamics trajectories: analysis of 2.2-ns trajectories of YPGDV, Biochemistry, 1993, 32, 412–420 CrossRef CAS.
- H. W. Van den Hooven, F. M. Lagerwerf, W. Heerma, J. Haverkamp, J. C. Piard, C. W. Hilbers, R. J. Siezen, O. P. Kuipers and H. S. Rollema, The structure of the lantibiotic lacticin 481 produced by Lactococcus lactis: Location of the thioether bridges, FEBS Lett., 1996, 391, 317–322 CrossRef.
- M. Van de Kamp, L. M. Horstink, H. W. van den Hooven, R. N. H. Konings, C. W. Hilbers, A. Frey, H. G. Sahl, J. W. Metzger and F. J. M. Vandeven, Sequence analysis ny NMR spectroscopy of the peptide lantibiotic epilancin K7 from Staphylococcus epidermidis K7, Eur. J. Biochem., 1995, 227, 757–771 CrossRef CAS PubMed.
- J. L. Parisot, S. Carey, E. Breukink, W. C. Chan, A. Narbad and B. Bonev, Molecular mechanism of target recognition by subtilin, a class I lanthionine antibiotic, Antimicrob. Agents Chemother., 2008, 52, 612–618 CrossRef CAS PubMed.
- J. R. Van der Meer, J. Polman, M. M. Beerthuyzen, R. J. Siezen, O. P. Kuipers and W. M. De Vos, Characterization of the Lactococcus lactis nisin A operon genes nisP, encoding a subtilisin-like serine protease involved in precursor processing, and nisR, encoding a regulatory protein involved in nisin biosynthesis, J. Bacteriol., 1993, 175, 2578–2588 CAS.
- O. P. Kuipers, H. S. Rollema, W. M. de Vos and R. J. Siezen, Biosynthesis and secretion of a precursor of nisin Z by Lactococcus lactis, directed by the leader peptide of the homologous lantibiotic subtilin from Bacillus subtilis, FEBS Lett., 1993, 330, 23–27 CrossRef CAS.
- C. J. Giffard, H. M. Dodd, N. Horn, S. Ladha, A. R. Mackie, A. Parr, M. J. Gasson and D. Sanders, Structure–Function Relations of Variant and Fragment Nisins Studied with Model Membrane Systems, Biochemistry, 1997, 36, 3802–3810 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra09897h |
| ‡ Current address: School of Pharmacy, University of Nottingham, Nottingham, NG7 2UH, UK. |
| This journal is © The Royal Society of Chemistry 2014 |