
Synthesis of spherical yttrium aluminum garnet via a mixed solvothermal method
Abstract
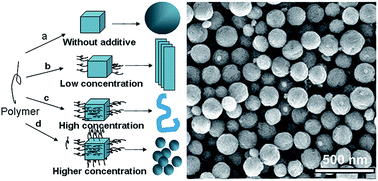
Yttrium aluminium garnet (YAG) powders were synthesized by a mixed solvothermal method. The crystal structure of the YAG powders was characterized by transmission electron microscopy (TEM), scanning electron microscopy (SEM), Fourier transform infrared spectroscopy (FT-IR) and X-ray diffraction (XRD). The impacts of the mixed solvents, the synthesis temperature and the polymer additive on the spherical grain formation and the crystal structure of the YAG has been researched by changing the synthetic conditions. The results show that spherical YAG powders were obtained using ethylenediamine–ethanol as mixed solvents with polyethylene glycol as additive at 220 °C for 20 h. Pure spherical YAG particles of 100–200 nm in diameter are formed directly in the process of mixed solvothermal reaction. The degree of crystallization and the crystal structure of the YAG phase have been improved with the increase of the synthesis temperature. The particles tend to be spherical and the distribution of particle size is homogeneous as expected. Finally, in this paper we investigated the formation mechanism of spherical YAG particles.
 Please wait while we load your content...
Please wait while we load your content...

