DOI:
10.1039/C4RA09867F
(Paper)
RSC Adv., 2014,
4, 57935-57944
Destruction mechanism of core–shell particles in impact polypropylene copolymer during short molten-state annealing†
Received
5th September 2014
, Accepted 29th October 2014
First published on 30th October 2014
Abstract
Morphology evolution of the dispersed phase with a multilayered core–shell structure in impact polypropylene copolymer (IPC) during molten-state annealing was systematically studied through scanning electron microscopy (SEM), phase contrast microscopy (PCM) and dynamic rheological test. To demonstrate the evolution path of the dispersed phase comprised of ethylene-propylene random copolymer (EPR) and ethylene–propylene block copolymer (EbP) during annealing, different binary blends comprised of different fractions were prepared and their diffusion behavior during liquid–liquid phase separation was investigated. Compared with EPR, EbP presented a higher diffusion rate in propylene homopolymer (hPP) matrix, owing to its lower molecular weight and lower entanglement density. The statistical results of EbP and EPR domain sizes reveal that the coalescence of EbP is faster than that of EPR. In addition, the interaction parameters of EbP/hPP and EbP/EPR estimated using the Nishi–Wang equation show that EbP has a stronger affinity for hPP than EPR. Based on the diffusion rates, entanglement densities of components and great disparity in viscosity between EPR and hPP, a potential mechanism was proposed for the morphology evolution of core–shell dispersed particles in IPC during molten-state annealing.
1 Introduction
Impact polypropylene copolymer (IPC) is an important commercial polyolefin used in many applications due to its excellent impact properties, especially in low temperatures.1–4 It is usually produced by a two-step polymerization involving bulk polymerization of propylene and subsequent gas-phase copolymerization of ethylene and propylene.5–8 Due to its special polymerization and complex composition, IPC can also be denoted as a multi-phase and multi-component polymer alloy.9,10
IPC mainly contains three components, i.e., ethylene–propylene random copolymer (EPR), a series of ethylene propylene block copolymers with different sequence lengths (EbP) and propylene homopolymer (hPP), and maybe a small amount of ethylene homopolymer (hPE).11–15 In our previous papers,5,16 we proposed a multilayered core–shell dispersed phase structure model for IPC. In this model, the dispersed phase is composed of an EbP core and two layers with the outer layer, EbP and the inner one, EPR. In particular, some EbPs pass through the EPR layer and form some bridges connecting the core and EbP layer. Some other researchers10,17,18 also demonstrated the similar core–shell dispersed phase structure except that there may be a little hPE in the core. EbP component plays a role as compatibilizer due to its good compatibility with both EPR and hPP components.5,19 Interestingly, the structure of dispersed phase can be spontaneously reconstructed in a certain condition by solution casting.10,17,18 The EbP component plays an important role in rebuilding the core–shell dispersed phase structure.19
The extraordinary properties of IPC have been revealed.10,18,20–22 Song et al.10 proposed that PE segments can gather together to form stable nuclei and play a role in increasing the crystallization rate of IPC by promoting the nucleation rate. Through a comparison study,3 we revealed that poor interfacial adhesion between the dispersed phase and the continuous matrix, larger dimensions and non-uniform distribution of dispersed phases are main reasons for the low impact toughness of those IPCs with a low content of ethylene–propylene segmented copolymer.
Similar to most of multicomponent polymer materials, the phase structure of IPC is also unstable during processing and the coarsening of phase domains occurs. Our previous work has studied the influence of long time annealing on the phase morphology of IPC.23 The influence of destroyed core–shell particles resulting from annealing on the crystallization behavior and the phase inversion phenomenon were revealed. However, up to now, there is no work to document the evolution path of morphology from the original core–shell structure to the common coarsening structure for IPC. As a result, the mechanism of the destruction of core–shell particles is still unclear. Since the complex phase morphology of IPC forms due to the fact that it is an in situ polymer alloy, the cause of the formation of core–shell particles is not revealed. Thus, it is necessary to study the destruction mechanism of these core–shell particles since it can be helpful to understand the reason for the formation of these core–shell particles. In this work, morphology evolution of core–shell dispersed particles in IPC during short molten-state annealing was studied through scanning electron microscope (SEM), phase contrast microscope (PCM), rheometry and differential scanning calorimetry (DSC), and a potential destruction mechanism of core–shell particles was proposed.
2 Experimental
2.1 Materials and sample preparation
The commercial IPC (SP179, molar percentage of ethylene component is about 13.5%) was purchased from SINOPEC Qilu Corporation Ltd., China. IPC was fractionated through temperature-gradient extraction fractionation method and n-octane was used as the solvent to successively extract the sample at different temperatures.5,23 25 g raw IPC resin was first completely dissolved in boiling n-octane. Subsequently, the solution was cooled down to 50 °C and held for 72 h, and the fraction was collected as EPR fraction.24 Then, the remaining sample was extracted at 100 °C for 72 h and the collected fraction was named as EbP fraction.24 After the above two-step extraction, the remainder was collected as the third fraction, hPP fraction.24 The molecular weights of IPC and the fractions were determined by gel permeation chromatography (GPC) at 150 °C. The GPC system used is a PL-GPC220 equipped with three PLgel 10 mm MIXED-B columns using polystyrene as standards and 1,2,4-trichlorobenzene (1.0 mL min−1) as the eluent. The results are listed in Table 1.
Table 1 Molecular weight and fraction amount for IPC and its fractions
| Sample |
Fraction amount (%) |
Mn × 104 |
Mw × 105 |
PD |
| IPC |
— |
4.39 |
1.74 |
3.96 |
| EPR |
18.96 |
8.45 |
2.27 |
2.68 |
| EbP |
11.30 |
1.60 |
1.80 |
11.25 |
| hPP |
69.74 |
4.60 |
1.51 |
3.28 |
Three blend samples, EbP/hPP, EbP/EPR and EPR/hPP were prepared by solution-mixing method, respectively. The fractions were first weighted according to the designated proportion as shown in Table 2, and then they were dissolved in boiling n-octane. Subsequently, the solution was stirred thoroughly for 30 min to ensure complete mixing. Finally, the solution was mixed with excessive methanol to participate. After filtration, the blend samples were obtained and dried at ambient temperature for 48 h and subsequently dried in vacuum at 80 °C for 24 h.
Table 2 Components weight for three binary blends
| Component |
EbP/hPP |
EbP/EPR |
EPR/hPP |
| EPR |
— |
18.96 g |
18.96 g |
| EbP |
11.30 g |
11.30 g |
— |
| hPP |
69.74 g |
— |
69.74 g |
2.2 SEM observation
IPC pellets were first annealed at 210 °C on a hot stage under nitrogen atmosphere for different time. And then fracture surface of pellets specimens obtained at liquid nitrogen was etched in 50 °C n-octane for 4 h, and observed using SEM (S-4800, Hitachi, Japan) after being coated with gold–palladium. The operating voltage was 3 kV and the magnification was 2000.
2.3 PCM observation
IPC pellets, EbP fraction and three binary blends containing EbP/hPP, EbP/EPR, EPR/hPP were observed using PCM (BX51, Olympus) which was equipped with an Olympus camera and the temperature was monitored with a temperature controlled hot stage (THMS600, Linkam Co.). The specimens were films with thickness of about 150 μm and sandwiched between two microscope cover slips for observation. PCM micrographs of the five specimens annealed at 210 °C for different time were taken as the morphology evolved. All thermal treatments proceeded under nitrogen atmosphere.
2.4 Dynamic rheological test
The dynamic rheological tests were conducted on an ARES (TA Instruments Corporation, USA) with parallel plate geometry of 25 mm diameter under air condition. The tests were performed under conditions as follows. (i) Isothermal frequency sweeps from 100 to 0.1 rad s−1 for hPP fraction, and 100 to 0.01585 rad s−1 for other two fractions and IPC. (ii) Isothermal shear rate sweeps from 0.001 s−1 to 10 s−1 for all samples. All tests were conducted at 210 °C and a strain of 5% which was ensured within the linear viscoelastic range was applied for frequency sweeps. The test specimens adding with a small amount of antioxidant (Irganox 1010) were prepared by compression molding at 180 °C under 10 MPa for 8 min.
2.5 DSC and thermal gravimetric analysis measurements
The thermal behavior was examined by using a Q100 DSC (TA Instruments Corporation, USA) with nitrogen as purge gas. The samples were first heated to 190 °C and held for 5 min to eliminate previous thermal history and then cooled to 40 °C and maintained for 5 min, and subsequently heated to 190 °C again. The later heating test was used to determine the melting point (Tm) and crystallinity. Both the heating and cooling rates were 10 °C min−1.
Thermal gravimetric analysis (TGA) was carried out by Pyris 1 TGA (Perkin-Elmer, USA) at 210 °C under nitrogen and air atmosphere.
3 Results and discussion
3.1 Phase morphology evolution of IPC during static annealing
Fig. 1 gives the SEM micrograph of the fracture surface of raw IPC after being etched by n-octane and its phase structure model. The detailed descriptions about the phase structure for IPC have been documented in our previous work.5 It is believed the dispersed phase can present three different cases depending on the degree of wrapping EbP core in the EPR layer: (i) isolated holes marked as ‘A’, (ii) the holes but containing a smaller granule marked as ‘B’, and (iii) aggregate granules marked as ‘C’. It is seen that the actual structure can be well described by the dispersed particle model. It is clearly visible that the EbP bridges connecting inner core and EbP layer pass through the EPR layer (seen in Fig. S1, ESI†). In addition, in the case of bridges only exist on the other side away from the viewer, an isolated EbP granule is left behind marked as ‘B′’ by removing the outer EPR layer.
 |
| | Fig. 1 Phase structure model of IPC and SEM micrograph of dispersed phase. | |
Fig. 2 presents the images of phase evolution for IPC samples annealed at 210 °C over different time. From PCM images, it is seen that the phase domains get larger with the annealing time increasing. However, more details can be observed from SEM images. The aggregate granules in SEM images are EbPs according to the phase model. When etching temperature rises to 100 °C, the EbPs are moved and only isolated holes exist (shown in Fig. S2†). As shown in Fig. 2a (SEM image), the dispersed phase existing in IPC pellet is extremely uniform and the size of dispersed phase is about 1 μm. However, the core–shell structure of dispersed phase is completely destroyed as to the sample annealed for 10 min, and the EbP aggregates seem to grow as they become more obvious. This fact differs from the observation of Chen,25 in which the core–shell particles still maintain a discrete state annealed at 200 °C for 200 min. He ascribed the stable core–shell particles in their results to the interfacially-active block copolymer which can suppress the phase coarsening process.26–28 However, the melting points of EbP obtained by extraction fractionation of IPC used in our experiments5 are significantly lower than those in Chen's, indicating the sequence lengths of EbP in this case are more shorter. Considering the shorter sequence lengths of block copolymer which may inhibit the interfacial activation and higher annealing temperature in our case, the obvious EbP aggregates observed in our results are reasonable. With annealing time increasing, EbP granules gradually form a network structure throughout the whole material for samples annealed for 20 and 30 min as shown in Fig. 2, whereas the EPR holes grow more slowly as compared to EbP. These results indicate that EPR and EbP present different coarsening behaviors during static annealing.
 |
| | Fig. 2 SEM images and PCM micrographs of IPC samples annealed at 210 °C for different time. (a) 0 min, (b) 10 min, (c) 20 min, (d) 30 min. For SEM observation, fracture surface of pellets specimens was obtained at liquid nitrogen and then was etched in 50 °C n-octane for 4 h. | |
It is worth noting that oxidative degradation and crosslinking could be one of the reasons for the instability of IPC melt at high temperatures.29 Despite the protective atmosphere of nitrogen during SEM and PCM observations, it is necessary to probe possible thermal degradation and crosslinking. Fig. 3 shows the TGA curves of IPC in nitrogen and air atmosphere at 210 °C. For air atmosphere, the weight floats up slightly and then drops violently. As for nitrogen atmosphere, almost no weight loss occurs. Consequently, the thermal degradation of IPC can be ignored during the experimental period in the nitrogen atmosphere. Moreover, the IPC pellet annealed at 210 °C for 30 min in the nitrogen atmosphere were extracted in boiling n-octane by a Soxhlet extractor for 48 h. After extracting, nothing remained, indicating that no crosslinking happens. All these results indicate that nitrogen atmosphere can avoid the extra chemical change of IPC in our cases.
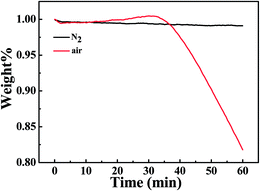 |
| | Fig. 3 TGA curves of IPC in nitrogen and air atmosphere at 210 °C. | |
3.2 Comparison of diffusion rates in different binary blends
Fig. 4 gives the morphology evolution of EbP/hPP, EPR/hPP and EbP/EPR blends annealed at 210 °C, respectively. The composition of each blend is coincident with weight ratio of components in the original IPC, i.e., EbP/hPP (11/70), EPR/hPP (19/70) and EbP/EPR (11/19). All three blends appear homogeneous at 0 min upon solution mixing, whereas different heterogeneities can be observed for all samples annealed for 40 min, indicating that phase separation takes place. In particular, the phase separation of EbP/hPP blend is more distinct than other two samples because of larger domains size, and the EPR/hPP sample only exhibits a slight heterogeneity. All samples coarsen gradually and their domains size becomes larger over time. When the annealing time gets to 240 min, the EbP/hPP sample appears a clear phase-separated morphology, but the EPR/hPP and EbP/EPR samples only present slight heterogeneity, indicating they present obviously different coarsening rates. In the case of phase separation of EPR/hPP, it has been confirmed to follow the spinodal decomposition (SD) mechanism30,31 and we have also found that it has a very slow diffusion rate.23 However, considering the morphology similarity between EPR/hPP and EbP/EPR as well the characteristic of morphology evolution of EbP/hPP, it is reasonable to claim that both EbP/hPP and EbP/EPR samples follow the SD mechanism. The growth rate of phase domains can be an indicator of diffusion rate of component in the blend. In EbP/hPP blend, hPP is the main component and EbP forms the dispersed phase according to the weight ratio listed in Table 2. And for other two blends, the main components are hPP and EPR, respectively. Consequently, the diffusion rates of fractions can be qualitatively compared as follows. (i) The diffusion rate of EbP in hPP matrix is comparatively fast. (ii) The diffusion rates of EPR in hPP matrix and of EbP in EPR matrix are similar and quite slow.
 |
| | Fig. 4 Phase contrast micrographs of samples annealed at 210 °C for different time. (A) EbP/hPP, (B) EPR/hPP, (C) EbP/EPR. Suffix number 1 stands for 0 min, 2 stands for 40 min, 3 stands for 140 min and 4 stands for 240 min. | |
3.3 Interaction parameters of EbP/hPP and EbP/EPR
Diffusion rates of components are also related to the interaction parameters of polymers. In order to study further, interaction parameters of EbP/hPP and EbP/EPR were estimated using Nishi–Wang equation.32| |
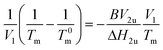 | (1) |
where the subscript 1 identified with the amorphous polymer and 2 with the crystalline polymer, V is the volume fraction, Vu is the molar volume of the repeating units, Tm is the melting point of mixture, T0m is the melting point of pure crystalline component and ΔH is the enthalpy of fusion per mole of repeating units. The interaction energy density characteristic of the polymer pair, B, can be obtained by the plot of quantity (1/Tm − 1/T0m)/V1 against V1/Tm. And then interaction parameter χ can be obtained by32| |
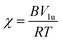 | (2) |
in which R is the gas constant, and T is the absolute temperature.
This equation has been widely applied to the amorphous/crystalline polymer blend. However, in EbP/hPP blend, the two components are both crystalline. In view of the condition used to deduce the equation that at the melting point of mixture the chemical potentials of the crystalline component in the crystalline and liquid phases should be identical,33 this equation is reasonable only in the melting region of mixture. Fig. 5a shows DSC curves for EbP, hPP and EbP/hPP mixtures. Due to two crystalline sequences, EbP shows the melting peaks of PE crystals and PP crystals, respectively. According to previous work,24 the melting peak at 115.5 °C is attributed to melting of PE crystals. Melting peaks at 136.0 °C and 144.6 °C are attributed to melting of PP crystals with low lamellae thickness. However, there is only a single melting peak for melting of PP crystals for every EbP/hPP mixture. The melting point (PP crystals) of mixtures as a function of the volume fraction of EbP and crystallinity of EbP and hPP are shown in Fig. 5b. Obviously, the crystallinity of EbP is far lower than that of hPP. Moreover, at melting region of EbP/hPP mixtures EbP is in the molten state. Consequently, EbP fraction can be regarded as amorphous component in melting region of EbP/hPP mixtures. This fact means that it may be reasonable to obtain interaction parameter of EbP/hPP by using Nishi–Wang equation. However, it should be pointed out that in this case we focus on the melting peak of PP crystals since hPP is regarded as crystalline polymer and shows only the single melting peak for PP crystals not for PE crystals.
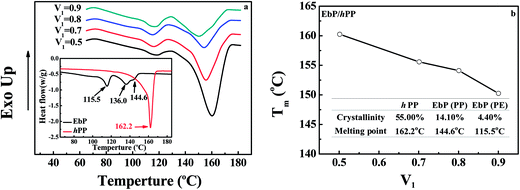 |
| | Fig. 5 (a) DSC curves of EbP, hPP and EbP/hPP mixtures, (b) melting points of EbP/hPP mixtures and crystallinity of hPP and EbP. V1 is volume fraction of EbP. Densities of EbP and hPP are 0.9 g cm−3 and 0.905 g cm−3, respectively. | |
The plots of quantity (1/Tm − 1/T0m)/V1 against V1/Tm of EbP/hPP and EbP/EPR are shown in Fig. 6. It should be pointed out that EbP is regarded as amorphous component for the system of EbP/hPP, and for EbP/EPR the EbP component is regarded as crystalline. Here, although two different polymer crystals exist in EbP,5 the melting point of PE crystals is used to deduce the interaction parameter of EbP/EPR system. The reason for this choice will be discussed in later.
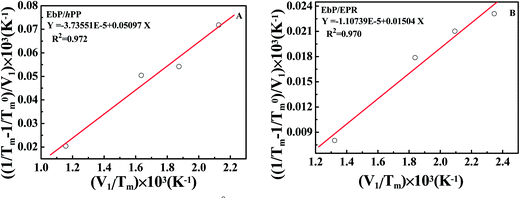 |
| | Fig. 6 A plot of the quantity (1/Tm − 1/T0m)/V1 against V1/Tm for EbP/hPP and EbP/EPR mixtures where Tm is the melting point of mixture, T0m is the melting point of pure crystalline component, and V1 is volume fraction of amorphous component. For EbP/hPP, T0m is used as 162.2 °C. For EbP/EPR, T0m is used as 115.5 °C. | |
The results can be acquired when the assumption is made to calculate Vu of EbP and EPR that the molar percentages of ethylene component in EbP and EPR are both about 13.5% and they are dispersed uniformly. The following values are used in the calculation: density of EPR is 0.854 g cm−3, ΔH of PP crystals is 7434 J mol−1,34 ΔH of PE crystals is 8201 J mol−1.35 However, it must be emphasized that ΔH of PE crystals for EbP should be multiplied by 0.135 since molecular chain of EbP consists of PE sequences and PP sequences. The results are as follow:
| χEbP-hPP = −0.1023 ± 0.0122 at 155 °C |
| χEbP-EPR = −0.0055 ± 0.0007 at 113 °C |
Since eqn (1) is reasonable only in the melting region of mixture, the interaction parameters for these two systems are at different temperatures. However, the interaction parameter obtained is an average value because EbP is a series of block copolymers with different sequence lengths. Anyway, both interaction parameters for the two systems are less than the critical χ that is the condition for the miscibility of the two polymers,36
| |
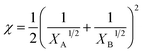 | (3) |
which is a positive value near zero. This fact indicates that there is a strong interaction between EbP and hPP, EbP and EPR also. However, it should be pointed out that the quantity (1/
Tm − 1/
T0m)/
V1 against
V1/
Tm does not show a linear relationship for EPR/hPP (seen in Fig. S3
†), meaning that there is no strong interaction between EPR and hPP. In view of the lower value of
χ, the interaction between EbP and hPP is much stronger than that between EbP and EPR at their respective temperatures. Moreover, the two-step polymerization results in the existing of a small amount of crystalline sequence lengths in EPR,
24 as shown in
Fig. 7. Taking the fact that EPR is the fraction dissolved in
n-octane at 50 °C into account, it is reasonable to conclude that the crystalline sequence in EPR fraction is probably ethylene segments rather than propylene segments. Being aware of the co-crystals between EbP and hPP,
5 it is supposed that co-crystals can also form in EbP/EPR blend. This is the reason for the choice of melting point of PE crystals to calculate the interaction parameter of EbP/EPR in
Fig. 6. Therefore, the strong interaction may origin from the existence of same segments for both EbP/hPP and EbP/EPR blends. We also used the melting points of PP crystals in EbP/EPR mixtures. However, the result does not show a linear relationship similar to that of EPR/hPP (seen in Fig. S3
†). Consequently, it is reasonable that the interaction between EbP and EPR comes from the same sequences in their molecular chains. Considering the small amount of crystalline sequence lengths in EPR, it has a weaker affinity for EbP. In other words, EbP has a stronger affinity for hPP than for EPR. At low temperatures, formation of co-crystals results in the existence of strong interaction between EbP and other two fractions. On the condition temperature rising, co-crystals disappear and phase separation can take place.
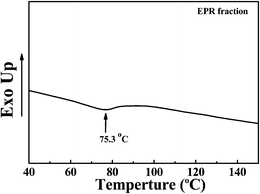 |
| | Fig. 7 DSC curve of EPR fraction. | |
3.4 Molecular dynamics
It seems to be contradictory that EbP/hPP has the lowest value of interaction parameter. However, the EbP/hPP blend displays the most pronounced coarsening behavior. It is well-known that whether the phase separation can take place at a given temperature is determined by thermodynamic factors. As indicated by the results in Fig. 4, all three blends are immiscible at 210 °C, so phase separation may take place at that temperature. The lower value of interaction parameter of EbP/hPP means that EbP has a stronger affinity for hPP than EPR. However, as to phase separation rate, it strongly depends on kinetic factors. It is believed that liquid–liquid phase separation is a process synchronously containing disentanglement and segment motion.37 And growth rate of phase domains depends strongly on the diffusion of macromolecular chains.38 On the other hand, macromolecular chains in phase domains should disentangle themselves before diffusing into the matrix. In order to study the reason for the most pronounced coarsening behavior in EbP/hPP blend, the molecular dynamics is worth being investigated. And in this section, entanglement densities, terminal relaxation times, viscosities and diffusion coefficients of EbP and EPR are investigated.
According to the tube model,39 entanglement molecular weight Me defined as the average molecular weight between entanglements can be deduced from the plateau modulus G0N. Fetters modified the tube model and the formula can be expressed as40
| |
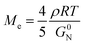 | (4) |
in which,
ρ is the density,
R is the gas constant and
T is the absolute temperature. The
Mn/
Me is defined as the “number of entanglements” per molecule according to Larson.
41 Here, the plateau modulus is deduced from the crossover modulus (
Gx, seen in Fig. S4
†) suggested by Wu.
42 And the terminal relaxation times (
τrep) are measured experimentally as the reciprocal of the frequency at which storage modulus is equal to loss modulus.
Table 3 gives the crossover modulus, plateau modulus, entanglement molecular weight, “number of entanglements” per molecule and terminal relaxation time for EPR and EbP. It can be seen that Mn/Me of EPR is much larger than that of EbP, meaning there are much more entanglements in the EPR domains. Moreover, τrep of EPR is almost 49 times as large as that of EbP. Both of them reveal that disentangling in EPR is harder than that in EbP. Considering the great disparity in entanglement density and terminal relaxation time between EPR and EbP, the result of pronounced coarsening behavior in EbP/hPP seems reasonable. However, it still needs more experimental evidences. Fig. 8 gives the complex viscosity (η*) and apparent viscosity (ηa) of IPC and its fractions at 210 °C. Test frequency (ω) for hPP ranges from 100 to 0.1 rad s−1 because of the instability resulting from too low viscosity in long time. In descending order of both complex viscosity and apparent viscosity, four specimens rank as EPR, IPC, EbP and hPP. The viscosity of EbP is significantly lower than that of EPR, indicating that EbP chain is easier to drag itself out of the droplet by resisting the viscous resistance, which also accords with the fact that EbP has a lower entanglement density than EPR.
Table 3 Entanglement molecular weight and terminal relaxation time for EPR and EbP at 210 °C
| |
Gx (×104 Pa) |
G0N (×105 Pa) |
Me (×103 g mol−1) |
Mn/Me |
τrep (ms) |
| EPR |
4.74 |
4.03 |
6.8 |
12.4 |
874.3 |
| EbP |
2.13 |
3.03 |
9.5 |
1.7 |
17.7 |
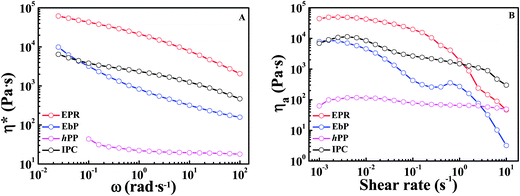 |
| | Fig. 8 (A) Frequency (ω) dependence of complex viscosity (η*) and (B) shear rate dependence of apparent viscosity (ηa) of IPC and its fractions at 210 °C. | |
The above results show EbP chain is easier to disentangle and drag itself out of the droplet and then diffuse into the matrix. Since the hPP matrix is determined, the diffusion coefficients of EbP and EPR can be semi-quantitatively compared supposing the influence of the same matrix is ignored. Based on the theory of entangled linear polymers,43,44 the diffusion coefficient (D) can be expressed as follows:
| |
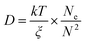 | (5) |
in which
k is boltzmann constant,
T is absolute temperature,
ξ is friction coefficient,
Ne is the number of Kuhn monomers in an entanglement strand and
N is the number of Kuhn monomers in a macromolecular chain. As a result, the ratio of diffusion coefficients for EbP and EPR can be obtained.
| |
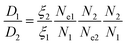 | (6) |
Due to the same matrix of hPP and EbP, EPR both contain ethylene and propylene monomers, it can be obtained roughly
Using the two equations
in which,
Mk is the molecular weight of Kuhn monomer,
Mn is molecular weight of macromolecular chain and
Me is entanglement molecular weight. Using the values of
Mn,
Me of EbP and EPR, we have
| |
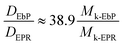 | (8) |
According to Rubinstein's works,44 we can get Mk-PP = 180 g mol−1 and Mk-PE = 150 g mol−1. Since EbP and EPR are both the copolymers of ethylene and propylene, we have
Mk-EbP, Mk-EPR ∈ (150, 180) g mol−1
Consequently,
As a result, it can be obtained
| |
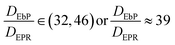 | (9) |
Eqn (9) tells us that in the same matrix, the diffusion rate of EbP is roughly 39 times than that of EPR, meaning the phase separation in EbP/hPP is much faster than that of EPR/hPP in accordance with the results shown in Fig. 4. Understandably, huge differences of molar mass, number of entanglements per molecule, terminal relaxation time and viscosity between EbP and EPR results in the great disparity in diffusion coefficient in the same hPP matrix. This result is also in keeping with the previous works that have also proved that larger molecular weight leads to small apparent diffusivity or small growth rate of periodic length in phase separation behavior of different polymer mixtures.45–48 Thus, despite EbP/hPP shows the lower interaction parameter phase separation takes place in a faster rate than EPR/hPP due to the kinetic factors.
3.5 Morphology evolution of core–shell dispersed phase in IPC
Fig. 9 gives PCM images of IPC and EbP samples annealed at 210 °C for different time. IPC sample at 0 min presents micro-phase separation, and phase domains coarsen with increasing annealing time. Phase domains are so clear at 40 min similar to that of EbP/hPP blend, indicating that diffusion rate of some component in IPC is comparatively fast. Considering the core–shell structure of dispersed phase in IPC, the morphology evolution of IPC may consist of the following three forms. (i) EbP gathers in hPP matrix. (ii) EPR gathers in hPP matrix. (iii) EbP gathers in EPR matrix. Nevertheless, the contributions of form (ii) and (iii) are considerably ignored due to too slow diffusion rates of EPR and EbP in the matrix of hPP and EPR, respectively. Moreover, the core–shell structure of dispersed phase can be destroyed in early 10 min as shown in Fig. 2. Thus, the large phase domains in IPC at 40 min are seemly contributed by gathering of EbP in hPP matrix.
 |
| | Fig. 9 Phase contrast micrographs of IPC and EbP samples annealed at 210 °C for different time. Suffix number 1 stands for 0 min, 2 stands for 40 min, 3 stands for 140 min and 4 stands for 240 min. | |
However, in view of the fact that EbP is a series of ethylene-propylene block copolymers with different sequence lengths, phase separation may take place in EbP itself. Fig. 9 also shows PCM images of EbP. Actually, micro-phase separation in EbP can be indeed seen. However, with increasing annealing time, phase domains hardly coarsen even at 240 min. This fact indicates that EbP can not form large domains because of the chemical covalent bond between ethylene and propylene units. Consequently, it seems reasonable that coarsening of EbP in hPP matrix leads to large phase domains in IPC.
Fig. 10 presents the size of EbP and EPR domains during annealing period. The results were obtained by counting the size of dispersed phase in SEM images. However, size of EPR domains was obtained from the SEM images of the samples after being etched by n-octane at 100 °C so as to eliminate the interference of EbP. Results reveal that EbP domains grow much faster than that of EPR. And the growth rates of phase domains can be obtained from the slope of straight lines fitted by size and annealing time. Considering the fact that EbP possesses a lower weight fraction (11%), the much higher growth rate in phase size is a good evidence for the faster kinetics of EbP.
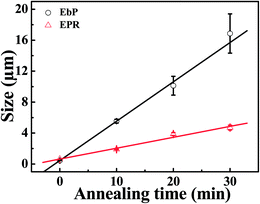 |
| | Fig. 10 Dispersed phase size of IPC samples annealed at 210 °C for different time. | |
Taking the above discussion into account, the morphology evolution of core–shell dispersed phase structure may be conducted in the following manner. During annealing period, enrichment of EbP in EPR layer is restricted due to the high viscosity of EPR. Compared with EPR, EbP have a stronger affinity for hPP due to the relatively more PP segments.24 Consequently, by the bridges connecting the inner EbP core and outer EbP layer, EbP core passes through the middle EPR layer and gets into hPP matrix. And then EbP gathers with a fast rate in hPP matrix, resulting in large phase domains in IPC in a short period of time. However, EPR gathers in hPP with a slow rate similar to the common EPR/hPP binary blend. In a word, it is different for EbP and EPR in the evolution of phase structure in IPC, and the schemes are plotted in Fig. 11.
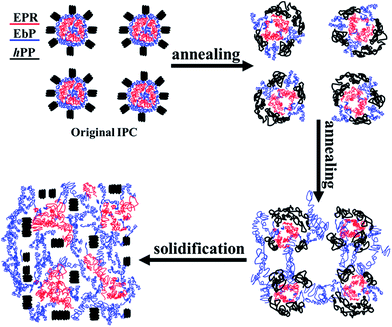 |
| | Fig. 11 Schematic model depicting morphology evolution of core–shell dispersed phase structure. | |
When temperature falls down, the phase structures at different stages depicted in Fig. 11 are frozen and preserved. Because of entrance into hPP matrix, more co-crystals between EbP and hPP can form, resulting in difficulty in being removed with EPR layer by being etched in n-octane at 50 °C. This fact indicates that the number of EbP aggregate granules on the fracture surface will become larger with increasing annealing time, which accords nicely with the results shown in Fig. 2. And the impact properties of IPC after being annealed have also been studied. The results reveal that impact strength shows a huge drop at the first 10 min and further decreases with increasing annealing time (shown in ESI†). This fact is reasonable in view of the destruction of core–shell particles and the coarsening of rubber domains. However, which one of the two factors, i.e., core–shell particles or rubber size, contributes the excellent toughness to IPC has been not clear. And the issue will be studied in our further work.
4 Conclusions
The multilayered core–shell structure of dispersed particles in IPC is unstable and will be completely destroyed during annealing at 210 °C based on the SEM images. Through online observation of PCM, the diffusion rates of three different blends were studied. As a result, the large phase domains in IPC during annealing are mostly contributed by the enrichment of EbP in hPP matrix due to its higher diffusion rate. As for EbP itself, only micro-phase separation can be observed. Huge difference of molar mass and entanglement density between EbP and EPR results in the great disparity in diffusion coefficient. The statistical results of EbP and EPR domains size reveal that enrichment of EbP is faster than that of EPR. Due to the lower viscosity of hPP, it is believed that hPP is indeed possible to be as a channel for the enrichment of EbP. By using Nishi–Wang equation, the interaction parameters of EbP/hPP and EbP/EPR were calculated, indicating that EbP has a stronger affinity for hPP than EPR. During annealing period, the inner EbP core can pass through the middle EPR layer by the EbP bridges connecting the core and outer shell and enter into hPP matrix, then gathering with a fast rate. The enrichment of EbP brings up the large phase domains in IPC in a short period of time, whereas EPR gathers with a slow rate in the hPP matrix similar to the common binary blend of EPR/hPP.
Acknowledgements
This work was supported by National Nature Science Foundation of China (no. 51173157, 51173165) and the Fundamental Research Funds for the Central Universities (no. 2013QNA4048).
References
- F. M. Mirabella, Polymer, 1993, 34(8), 1729–1735 CrossRef CAS.
- W. Rungswang, P. Saendee, B. Thitisuk, T. Pathaweeisariyakul and W. Cheevasrirungruang, J. Appl. Polym. Sci., 2013, 128(5), 3131–3140 CrossRef CAS.
- H. S. Tan, L. Li, Z. N. Chen, Y. H. Song and Q. Zheng, Polymer, 2005, 46(10), 3522–3527 CrossRef CAS PubMed.
- T. McNally, P. McShane, G. M. Nally, W. R. Murphy, M. Cook and A. Miller, Polymer, 2002, 43(13), 3785–3793 CrossRef CAS.
- C. H. Zhang, Y. G. Shangguan, R. F. Chen, Y. Z. Wu, F. Chen, Q. Zheng and G. H. Hu, Polymer, 2010, 51(21), 4969–4977 CrossRef CAS PubMed.
- Z. H. Sun, F. S. Yu and Y. C. Qi, Polymer, 1991, 32(6), 1059–1064 CrossRef CAS.
- Y. Li, J. T. Xu, Q. Dong, Z. S. Fu and Z. Q. Fan, Polymer, 2009, 50(21), 5134–5141 CrossRef CAS PubMed.
- Z. Q. Fan, Y. Q. Zhang, J. T. Xu, H. T. Wang and L. X. Feng, Polymer, 2001, 42(13), 5559–5566 CrossRef CAS.
- Y. Chen, W. Chen and D. C. Yang, Polymer, 2006, 47(19), 6808–6813 CrossRef CAS PubMed.
- S. J. Song, J. C. Feng, P. Y. Wu and Y. L. Yang, Macromolecules, 2009, 42(18), 7067–7078 CrossRef CAS.
- H. N. Cheng and G. H. Lee, Macromolecules, 1987, 20(2), 436–438 CrossRef CAS.
- T. Hayashi, Y. Inoue, R. Chujo and T. Asakura, Polymer, 1988, 29(10), 1848–1857 CrossRef CAS.
- E. W. Hansen, K. Redford and H. Oysaed, Polymer, 1996, 37(1), 19–24 CrossRef CAS.
- Z. S. Fu, Z. Q. Fan, Y. Q. Zhang and L. X. Feng, Eur. Polym. J., 2003, 39(4), 795–804 CrossRef CAS.
- Q. Dong, X. F. Wang, Z. S. Fu, J. T. Xu and Z. Q. Fan, Polymer, 2007, 48(20), 5905–5916 CrossRef CAS PubMed.
- H. S. Tan, K. Xie, W. H. Liu, B. Hou, Y. G. Shangguan and Q. Zheng, Acta Polym. Sin., 2006, 9, 1106–1111 Search PubMed.
- Y. Chen, W. Chen and D. Yang, J. Appl. Polym. Sci., 2008, 108(4), 2379–2385 CrossRef CAS.
- S. J. Song, J. C. Feng and P. Y. Wu, Polymer, 2010, 51(22), 5267–5275 CrossRef CAS PubMed.
- C. Y. Tong, Y. Lan, Y. Chen, D. C. Yang and X. N. Yang, J. Appl. Polym. Sci., 2012, 123(3), 1302–1309 CrossRef CAS.
- F. Luo, C. L. Xu, K. Wang, H. Deng, F. Chen and Q. Fu, Polymer, 2012, 53(8), 1783–1790 CrossRef CAS PubMed.
- S. J. Song, P. Y. Wu, J. C. Feng, M. X. Ye and Y. L. Yang, Polymer, 2009, 50(1), 286–295 CrossRef CAS PubMed.
- F. Luo, C. L. Xu, N. Y. Ning, K. Wang, H. Deng, F. Chen and Q. Fu, Polym. Int., 2013, 62(2), 172–178 CrossRef CAS.
- R. F. Chen, Y. G. Shangguan, C. H. Zhang, F. Chen, E. Harkin-Jones and Q. Zheng, Polymer, 2011, 52(13), 2956–2963 CrossRef CAS PubMed.
- C. H. Zhang, Y. G. Shangguan, R. F. Chen and Q. Zheng, J. Appl. Polym. Sci., 2011, 119(3), 1560–1566 CrossRef CAS.
- Y. Chen, W. Chen and D. Yang, Eur. Polym. J., 2007, 43(7), 2999–3008 CrossRef CAS PubMed.
- I. M. Lifshitz and V. V. Slyozov, J. Phys. Chem. Solids, 1961, 19(1–2), 35–50 CrossRef.
- D. W. Park and R. J. Roe, Macromolecules, 1991, 24(19), 5324–5329 CrossRef CAS.
- Y. Kobori, I. Akiba and S. Akiyama, Polymer, 2000, 41(15), 5971–5975 CrossRef CAS.
- Y. G. Shangguan, C. H. Zhang, Y. L. Xie, R. F. Chen, L. Jin and Q. Zheng, Polymer, 2010, 51(2), 500–506 CrossRef CAS PubMed.
- N. Inaba, K. Sato, S. Suzuki and T. Hashimoto, Macromolecules, 1986, 19(6), 1690–1695 CrossRef CAS.
- S. W. Lim, K. H. Lee and C. H. Lee, Polymer, 1999, 40(10), 2837–2844 CrossRef CAS.
- T. Nishi and T. T. Wang, Macromolecules, 1975, 8(6), 909–915 CrossRef CAS.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, 1953 Search PubMed.
- J. X. Li, W. L. Cheung and D. M. Jia, Polymer, 1999, 40(5), 1219–1222 CrossRef CAS.
- J. D. Hoffman and R. L. Miller, Polymer, 1997, 38(13), 3151–3212 CrossRef CAS.
- R. L. Scott, J. Chem. Phys., 1949, 17(3), 279–284 CrossRef CAS PubMed.
- Y. Lin, Y. G. Shangguan, M. Zuo, E. Harkin-Jones and Q. Zheng, Polymer, 2012, 53(6), 1418–1427 CrossRef CAS PubMed.
- Q. Zheng, M. Peng, Y. H. Song and T. J. Zhao, Macromolecules, 2001, 34(24), 8483–8489 CrossRef CAS.
- M. Doi and S. F. Edwards, The theory of polymer dynamics, Clarendon Press, Oxford, 1986 Search PubMed.
- L. J. Fetters, D. J. Lohse, D. Richter, T. A. Witten and A. Zirkel, Macromolecules, 1994, 27(17), 4639–4647 CrossRef CAS.
- R. G. Larson, T. Sridhar, L. G. Leal, G. H. McKinley, A. E. Likhtman and T. C. B. McLeish, J. Rheol., 2003, 47(3), 809–818 CrossRef CAS.
- S. Wu, J. Polym. Sci., Part B: Polym. Phys., 1989, 27(4), 723–741 CrossRef CAS.
- P. G. de Gennes, Scaling concepts in polymer physics, Cornell university press, 1979 Search PubMed.
- M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, 2003 Search PubMed.
- Y. Huang, M. Song and G. Cong, Acta Polym. Sin., 1991, 6, 678–683 Search PubMed.
- H. Matsuyama, T. Maki, M. Teramoto and K. Asano, J. Membr. Sci., 2002, 204(1–2), 323–328 CrossRef CAS.
- R. Gelles and C. W. Frank, Macromolecules, 1983, 16(9), 1448–1456 CrossRef CAS.
- M. Takenaka, T. Izumitani and T. Hashimoto, Macromolecules, 1987, 20(9), 2257–2264 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra09867f |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 




















