
Changes in the supramolecular structures of cellulose after hydrolysis studied by terahertz spectroscopy and other methods†
Hong Guoa,
Mingxia Heb,
Renliang Huangc,
Wei Qiad,
Weihua Guoa,
Rongxin Su*ad and
Zhimin Hea
aState Key Laboratory of Chemical Engineering, Tianjin Key Laboratory of Membrane Science and Desalination Technology, School of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, P. R. China. E-mail: surx@tju.edu.cn; Fax: +(86)022-27407599; Tel: +(86)022-27407599
bCollege of Precision Instrument and Optoelectronics Engineering, Tianjin University, Tianjin 300072, China
cSchool of Environmental Science and Engineering, Tianjin University, Tianjin 300072, China
dCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300072, China
First published on 17th October 2014
Abstract
Hydrogen bonding is one of dominant forces in crystalline cellulose and lignocellulose, however, it is still a big challenge to evaluate the changes in the hydrogen bonding strength. Here, we reported a new method for measuring the changes in the hydrogen bonding strength of cellulose-based materials by terahertz time domain spectroscopy (THz-TDS). Avicel, corncob and their residual substrates after enzymatic hydrolysis were chosen as the targeted cellulose to demonstrate this method. THz adsorption in the range 0.5–2.5 THz and refractive index in the range 0.5–1.0 THz provided a direct signal corresponding to an increase in the hydrogen bonding strength for the residual samples (Avicel and corncob) after enzymatic hydrolysis. The THz results were further compared with those obtained from X-ray diffraction (XRD) and Fourier transform infrared spectroscopy (FTIR) analysis. As a quick and non-invasive technique, THz spectroscopy provides unique information about the changes in the hydrogen bonding strength of cellulose-based materials. Therefore, it is a promising way to directly evaluate the hydrogen bonding strength of cellulosic macromolecules.
Introduction
Lignocellulose, the most abundant and renewable biomass, has a great potential to serve as a sustainable supply of fuels and chemicals. In recent years, a large number of research efforts have focused on the bioconversion of lignocellulose into biofuels, such as ethanol and butanol.1–3 During this process, effectively breaking the recalcitrant structure and converting the polysaccharides to fermentable sugars are the major barriers in the commercialization of cellulosic biofuels. The recalcitrance of the crystalline cellulose in biomasses to enzymatic hydrolysis is mainly because of the strong intermolecular interactions, including hydrogen bonding and van der Waals force. The hydrogen bonds within the cellulose molecule and the adjacent cellulose chains provide a big barrier for cellulose deconstruction and enzymatic hydrolysis.4–11 To better understand the enzyme–substrate interactions and to improve the process of enzymatic hydrolysis, it is essential to measure the changes in the hydrogen bond strength, which is the accumulated strength of hydrogen bond, during the hydrolysis process.Fourier transform infrared spectroscopy (FTIR) shows a high sensitivity to various conformations and microenvironment of chemical groups such as hydroxyl and glycosidic bonds present in cellulose chains, Therefore, it has often been used to evaluate the hydrogen bonding strength in cellulosic substrates.12 In previous studies, the ratio of the absorbance at some characteristic peaks were introduced as the empirical hydrogen bonds intensity (or the degree of destruction), such as the absorbance ratios of A4000–2995/A1337, A1430/A897, and A897/A2900.10,13–15 However, the other components (e.g., lignin, hemicellulose) in lignocellulose also have the similar chemical groups (e.g., OH, C–H) that significantly influence the IR adsorption of characteristic groups derived from cellulose and thus renders a challenge to obtain exact hydrogen bonding strength. Deuterium exchange coupled with FTIR was used to monitor the structural change of lignocellulose during various treatments by following the change in the retention of the OD group at around 2500 cm−1 in the FTIR spectrum, including cellulose accessibility in aqueous systems and irreversible microfibril aggregation.16,17 Moreover, X-ray diffraction (XRD) is another auxiliary technique to evaluate the change in the hydrogen bonds within cellulose-based materials by detecting the amount of crystalline cellulose, which possesses strong hydrogen bonds.7,9,10,18,19 The crystallinity index obtained from XRD represents the relative amount of crystalline cellulose in the lignocellulosic substrate. Therefore, XRD results cannot reflect the actual crystallinity of cellulose in the presence of amorphous components (hemicellulose and lignin) other than cellulose. Previous studies also demonstrated that the crystalline index from XRD yields contradictory results regarding its effect on the enzymatic hydrolysis in different substrate systems.1,8 As well known, the crystallinity of cellulose is directly related to the intra- and intermolecular hydrogen bonds. Therefore, it might be more accurate to employ hydrogen bonding strength to characterize the cellulose crystallinity.
Terahertz time domain spectroscopy (THz-TDS) is a powerful tool for analyzing the structural and constitutional changes in materials, because of its high signal-to-noise ratio and broad spectra band constituting individual THz pulse.20 For example, the THz-TDS technology has been successfully applied in monitoring the conformation transition of protein,21 in the detection of PCR-amplified DNA22 and antibiotic residues in food industry.23 In general, the absorption features in the terahertz region are dominated by the intermolecular vibrations, which are related to the movement of atoms and/or molecules. In particular, the low-frequency vibrations between 0.5 and 3.0 THz are dominated by non-covalent intermolecular interactions such as hydrogen bonds, electrostatic force, and Van der Waals force. Previous studies have demonstrated that THz-TDS is a promising technology to determine these weak interactions in purine, insulin, and inks.24–26 As a quick and non-invasive technique, THz-TDS provides unique information about the changes in weak interactions. As mentioned above, crystalline cellulose contains strong hydrogen bonds. However, to date, no report has discussed the utilization of THz-TDS in determining the hydrogen bonding strength in lignocellulosic substrates.
In this study, we introduced the application of THz-TDS in evaluating the hydrogen bonding strength of cellulose. Avicel and corncob were chosen as the target substrates, because they are known to contain crystalline cellulose. Moreover, we also analyzed the changes in the hydrogen bonding strength of their residual solids after enzymatic hydrolysis. To further analyze the THz results, these cellulosic samples were characterized using XRD and FTIR techniques, to obtain the complementary structural information. In addition, we also discuss the results from size-exclusion chromatography with multi-angle laser light scattering (SEC-MALLS), as reported in our previous studies.27,28
Materials and methods
Materials
Microcrystalline cellulose (Avicel PH101) was purchased from Sigma (MO, USA). Air-dried corncob was collected from a local farm (Tianjin, China), pre-milled and screened to a nominal size of 20–80 meshes. Cellulase (Spezyme CP) derived from Trichodermareesei was a kind gift from Genencor International (CA, USA) in 2009. β-Glucosidase (Novozyme 188) was purchased from Sigma (St. Louis, MO, USA). The average activities of Spezyme CP and Novozyme 188 were 117 filter paper unit (FPU) mL−1 and 926 cellobiase unit (CBU) mL−1, respectively. All other reagents were of analytical grade and were obtained from commercial sources.Samples preparation
Corncob was soaked in aqueous ammonia (15 wt%) at a solid-to-liquid ratio (w/v) of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6. The solid/liquid slurry was then incubated in a water bath at 60 °C for 6 h without any agitation. After treatment, the corncob was separated by filtering and washed with distilled water to remove the residual ammonia until the pH became neutral. The resulting solid was oven-dried at 100 °C for 12 h and collected for subsequent experiments. The carbohydrate and lignin contents were determined by following the National Renewable Energy Laboratory (NREL) chemical analysis and standard testing procedure.
:6. The solid/liquid slurry was then incubated in a water bath at 60 °C for 6 h without any agitation. After treatment, the corncob was separated by filtering and washed with distilled water to remove the residual ammonia until the pH became neutral. The resulting solid was oven-dried at 100 °C for 12 h and collected for subsequent experiments. The carbohydrate and lignin contents were determined by following the National Renewable Energy Laboratory (NREL) chemical analysis and standard testing procedure.
To collect the residual cellulosic samples, Avicel and pretreated corncob were added into the citrate buffer (50 mM, pH 4.8) solution at a solid loading of 5% (w/v). The enzyme loadings for Avicel were 40 FPU g−1-glucan Spezyme CP, 60 CBU g−1-glucan Novozyme 188, whereas 30 FPU g−1-glucan and 30 CBU g−1-glucan for corncob. The enzymatic hydrolysis was carried out in an air shaker at 150 rpm and at 50 °C. After enzymatic hydrolysis, the soluble sugars in the supernatant were determined by high-performance liquid chromatography (HPLC) using an Aminex HPX-87H column (Bio-Rad, Hercules, CA, USA). All the solid residues were separated and incubated in a citrate buffer (50 mM, pH 7.0) with a papain–trypsin (2 mg/2 mg) mixture to remove the bound enzymes. These reactions were carried out at 50 °C with constant shaking for 12 h; and the solids were filtered out, washed with distilled water three times and, thereafter, oven-dried for the successive analysis.
Terahertz time domain spectroscopy (THz-TDS)
The THz measurements were performed at room temperature in a photoconductive switch-based terahertz time-domain system, which consists of four parabolic mirrors in an 8-F confocal geometry, with a useful bandwidth of 0.1–2.5 THz and an amplitude signal-to-noise ratio greater than 1.5 × 104:1. The 8-F confocal system compressed the terahertz beam to a frequency-independent diameter of 3.5 mm, and the THz radiation was excited by optical pulses from a mode-locked Ti-sapphire laser (λ = 800 nm, TFWHM = 25 fs). The cellulosic samples were naturally placed, without applying additional pressure, in a silicon cell composed of two parallel, 0.64 mm spaced and 0.64 mm thick windows. An identical empty cell was used as a reference during the data acquisition.
X-ray powder diffraction (XRD)
The XRD measurements were performed on an X'Pert Pro X-ray diffractometer (PANalytical, Holland). The diffracted intensity of the Co Kα radiation (λ = 1.789 Å, 30 kV, 30 mA) was measured in a 2θ range between 5° and 50° at the scan rate of 12° min−1. The crystallinity index (CrI) was calculated according to the method adopted by Segal et al.29 The formula used for the above calculation is as follows:
 | (1) |
Fourier transform infra-red (FTIR)
The FTIR spectra were recorded on a Nicolet-560E.S.P FTIR (Thermal Nicolet Co, USA) with a KBr pellet method in the range of 400–4000 cm−1. A total of 16 scans were accumulated with a resolution of 4 cm−1 for each spectrum. The relative absorbance (Arλ) for the band λ attributed to the hydrogen bonds was calculated using the C–H stretching band (2900 cm−1 for Avicel, 2920 cm−1 for corncob) as an internal standard, according to the empirical formula described in a previous study.13 The formula used for this calculation is as follows:
 | (2) |
Results and discussion
Changes in the chemical structure upon hydrolysis
The chemical compositions of these test materials, as a key factor influencing hydrogen bonds, were determined in this work. For Avicel, after 48 h of enzymatic hydrolysis, approximately 20% of the initial cellulose was hydrolyzed to glucose, leaving the residual cellulose for subsequent analysis. For corncob, enzymes catalyzed the hydrolysis of glucan and xylan, as shown in Fig. 1. Within the first 12 h, the glucan content in corncob significantly decreased from 45.79–18.4%, whereas the xylan content decreased from 37.01–18.4%. Subsequently, a slow degradation was observed up to 48 h. | ||
| Fig. 1 The contents of glucan and xylan (based on the initial dry matter) in the residual corncob during the enzymatic hydrolysis. | ||
Table 1 summarizes the main components of corncob before and after the 48 h enzymatic hydrolysis. The relative content of the glucan in the residual corncob was just 20.1%, whereas the xylan and lignin were 37.5% and 42.4%, respectively. In other words, approximately 10%, 23% and 88% of the initial glucan, xylan and lignin, respectively, were retained in the residual corncob. In terms of Avicel, no evident change in the glucan and hemicellulose contents was found after enzymatic hydrolysis (Table 1).
| Materials | Glucan (%) | Xylan (%) | Lignin (%) |
|---|---|---|---|
| Corncob | 45.79 | 37.01 | 11.14 |
| Residual corncob | 20.1 | 37.5 | 42.4 |
| Avicel | 96.2 | 3.8 | |
| Residual avicel | 96.5 | 3.5 |
XRD analysis
Prior to the evaluation of hydrogen bonds, we employed XRD to characterize the samples, aiming to investigate the structural change in the cellulosic substrates due to enzymatic hydrolysis. As shown in Fig. 2a, the initial and residual Avicel showed similar XRD spectra with similar crystallinity indices (CrI, 76.5% vs. 75.2%). There was no significant change in the relative crystallinity between the two samples. Although it is not suitable to estimate the amount of crystalline and amorphous cellulose in the lignocellulosic samples, the XRD peaks are useful for comparing the relative differences among the various cellulosic samples. The CrI values of Avicel in the previously studied reports were mostly shown in a wide range of 62–87.6%, using XRD peaks, whereas a recent research report even showed an extreme high value (higher than 90%).30 As reported in previously studied reports, no evident structural change in Avicel was observed under mild conditions.16 Here, we adopted the average value of three samples as the presenting CrI, and it showed consistency with the data of the references. In our previous study,28 using SEC-MALLS, we found that the residual Avicel had considerably higher average molecular weight than the original Avicel (Mw, 103 kDa vs. 59 kDa) and radii of gyration (Rg, 49.5 nm vs. 23.4 nm), indicating the presence of longer cellulose chains in the residual substrate. We thus speculated that the residual cellulose should have different hydrogen bond strengths than that in the initial cellulose. Unfortunately, this speculation could not be confirmed by XRD analysis.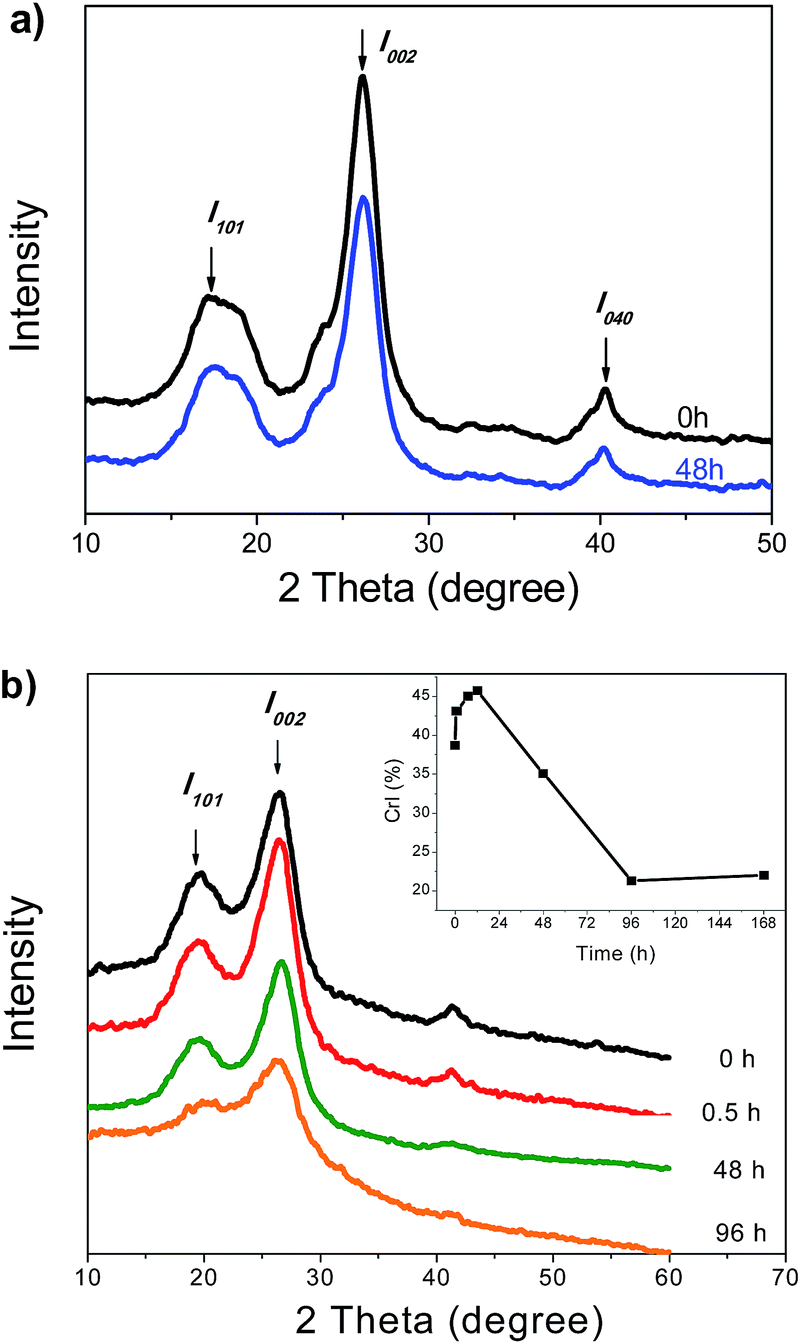 | ||
| Fig. 2 XRD profiles of Avicel (a) corncob (b) before and after the enzymatic hydrolysis. The inset in Fig. 2b shows the change in the crystalline index (CrI) of corncob during the enzymatic hydrolysis. | ||
The XRD patterns of corncob during the enzymatic hydrolysis are shown in Fig. 2b. The diffraction patterns of the samples shared similar shapes within 0–48 h, whereas a significant decrease in the intensity of the I002 peak was found at 96 h. The inset figure shows the change in the CrI, as calculated from the diffraction patterns. At the initial stage (0–0.5 h), the CrI increased rapidly from 38.7–43.1%, indicating an increased content of the crystalline components in corncob. Further increase in the CrI up to 45% was found in the following 11.5 h. The increased CrI should be mainly attributed to the hydrolysis of the disordered cellulose and hemicellulose. As the reaction proceeded, the CrI decreased to 35.1% at 48 h and to 22% at 168 h, suggesting that the crystalline cellulose was degraded or destructed gradually at this stage. The decrease in the glucan content of the residual corncob from 12–48 h (Fig. 1) was also in agreement with this deduction.
Previous studies had also demonstrated that the removal of amorphous components (e.g., hemicellulose and lignin) could result in an increased CrI of lignocellulosic substrates.31–34 However, the degradation of the disordered cellulose and hemicellulose has little effect on the ordered structure of the crystalline cellulose in the initial reaction.35 On the other hand, the hydrolysis of crystalline cellulose could lead to a decrease in CrI of the substrate, but it can't reflect the crystalline change in the residual cellulose of the substrate. Overall, although XRD is often used to determine the crystalline structure and crystallinity index of cellulosic substrates, it is difficult to determine the actual crystallinity and hydrogen bonding strength in cellulosic substrates, particularly in the presence of amorphous hemicellulose and lignin.36,37
THz spectroscopy analysis
The absorption coefficients and refractive indices of Avicel before and 48 h after the enzymatic hydrolysis in the range of 0.1–2.5 THz are shown in Fig. 3. It can be clearly seen that these two samples shared the similar spectral characteristics, lacking a prominent absorption peak, which was also observed in some other macromolecules, such as BSA and cytochrome c.38–40 However, they presented significantly different absorption intensity in the range of 1.5–2.5 THz. Specifically, the residual cellulose had an increased absorbance (e.g., from 13.5–18.8 at 1.5 THz and from 30.2–55.1 at 2.5 THz) in this spectral range. Fig. 3b shows the refractive indexes of the initial and residual Avicel in the range 0.1–2.5 THz. Both of them showed steady refractive indexes of 1.12 and 1.25, respectively. As shown in Fig. 3b, the residual cellulose presents a higher refractive index. Low frequency vibrational modes in THz-TDS were related to hydrogen bonding and other weak interactions.24–26 Particularly, a higher absorption and refractive index could be observed in self-assembled insulin nanofibers,25 as well as in purine and adenine at a lower temperature.24 As we know, both the self-assembly process and the decreased temperature could enhance the intermolecular interaction, such as π–π stacking and hydrogen bonding.41–43 Therefore, an increase in the absorption intensity for the residual cellulose should be regarded as a signal an enhancement of the intermolecular interactions (hydrogen bonding strength and van der Waals forces). In our previous study, the SEC-MALLS results suggested that the celluloses with a low molecular weight (Mw) were preferentially hydrolyzed to glucose, leaving behind the residual celluloses with a higher Mw.28 Based on these findings, we expected that the residual cellulose with higher absorption and refractive index value at low THz frequency should exhibit a stronger hydrogen bonding strength. This recalcitrant structure could thus hinder enzyme access to the residual cellulose for further hydrolysis, leading to a low cellulose conversion (∼20%). | ||
| Fig. 3 The absorption coefficient (a) and refractive index (b) of Avicel before (0 h) and after 48 h of enzymatic hydrolysis in the range 0.1–2.5 THz at 293 K. | ||
To further evaluate the hydrogen bonding strength in lignocellulose, we characterized the initial and residual corncob after 48 h of enzymatic hydrolysis by THz-TDS. The corncob showed a featureless absorption profile with monotonously increasing absorption at high frequencies (Fig. 4a), which is similar to that in the case of Avicel. Compared to the initial corncob, the residual corncob showed a higher absorbance in the range of 0.5–2.5 THz. Fig. 4b shows the change in the refractive index of corncob upon enzymatic hydrolysis. The refractive indexes of the initial and residual corncob were stabilized at about 1.24 and 1.20 in the range of 0.5–1.0 THz, respectively (as shown in the inset of Fig. 4b). A minor increase in the refractive index of the residual corncob at low frequency might be attributed to the removal of the low molecular-weight or disordered glucan with weak hydrogen bonding strength. A slight decrease in the refractive index of the residual corncob at a high frequency might be attributed to lignin accumulation upon enzymatic hydrolysis.44 In our previous study, we found that the cellulose in the residual corncob possessed a considerably higher Mw (1130 kDa) as compared to that (680 kDa) in the case of the initial corncob.27 The increased Mw indicated that some long chain celluloses in the residual substrate were still not attacked even after 48 h of hydrolysis, whereas the short chain cellulose was degraded preferentially in this process. According to the THz-TDS and SEC-MALLS results, the residual cellulose in corncob after the enzymatic hydrolysis should have a higher degree of polymerization and stronger hydrogen bonding strength compared to the original cellulose.
 | ||
| Fig. 4 The absorption coefficient (a) and refractive index (b) of corncob before (0 h) and after 48 h of enzymatic hydrolysis in the range 0.1–2.5 THz at 293 K. | ||
Based on the THz spectra from single cellulose and cellulose complex, we proposed that the adsorption in the range of 0.5–2.5 THz and the refractive index in the range of 0.5–1.0 THz could be used for evaluating the change in the hydrogen bonding strength of the cellulose materials during various processing treatments. The increased adsorption and refractive index indicated an increase in the hydrogen bonding strength.
FTIR analysis
To further confirm the feasibility of determining the hydrogen bonding strength using THz-TDS, we employed FTIR to characterize the samples to verify the above deduction.Fig. 5 shows the FTIR spectra of Avicel samples before and 48 h after the enzymatic hydrolysis over a wavenumber range of 4000–2800 cm−1 and 1800–600 cm−1. In general, the peaks at 3200–3600 cm−1 were related to the O–H stretching vibration, in which the adsorption at 3455–3410 cm−1, 3375–3340 cm−1, and 3310–3230 cm−1 was attributed to the intramolecular O2–H⋯O6, O3–H⋯O5, and intermolecular O6–H⋯O3 hydrogen bonds, respectively.45 As shown in Fig. 5a and S1,† after enzymatic hydrolysis, the peaks at 3455–3410 cm−1 and 3375–3340 cm−1 (derived from the residual Avicel), shifted to lower wavenumbers and showed a stronger adsorption, indicating an increase in the intramolecular hydrogen bonding strength within cellulose, whereas the peak related to the intermolecular hydrogen bonding showed a weak absorption, which might be resulting from the degradation of Avicel upon hydrolysis. On the other hand, the peak at 897 cm−1 originated from the β-glycosidic linkages associated with C1–H deformation and O–H bending. The bands 2920 cm−1, 1372 cm−1 and 1423 cm−1 were assigned to the C–H stretching in the methylene groups, aliphatic C–H stretching, and aromatic skeletal vibrations combined with C–H in plane deformations, respectively.46,47 According to the empirical formula, the absorbance ratio of the bands at 897 cm−1 (Ar897), 1375 cm−1 (Ar1375), and 1430 cm−1 (Ar1430) to 2915 cm−1 corresponded to the degree of hydrogen bonds destroyed.13 As shown in Table 2, these parameters for the residual Avicel decreased as compared to the original substrate. These results indicated that the hydrogen bonding strength in cellulose increased after enzymatic hydrolysis. The FTIR results were highly consistent with those obtained from the THz analysis. In addition, the characteristic adsorption peak of crystalline cellulose at 1430 cm−1 became weaker for Avicel after enzymatic hydrolysis, suggesting that the crystalline cellulose was partially degraded to glucose in this process. Based on the THz and FTIR results, it was evident that the enzymes could degrade part of cellulose but lead to an increase in the accumulated hydrogen bonding strength for the residual substrate.
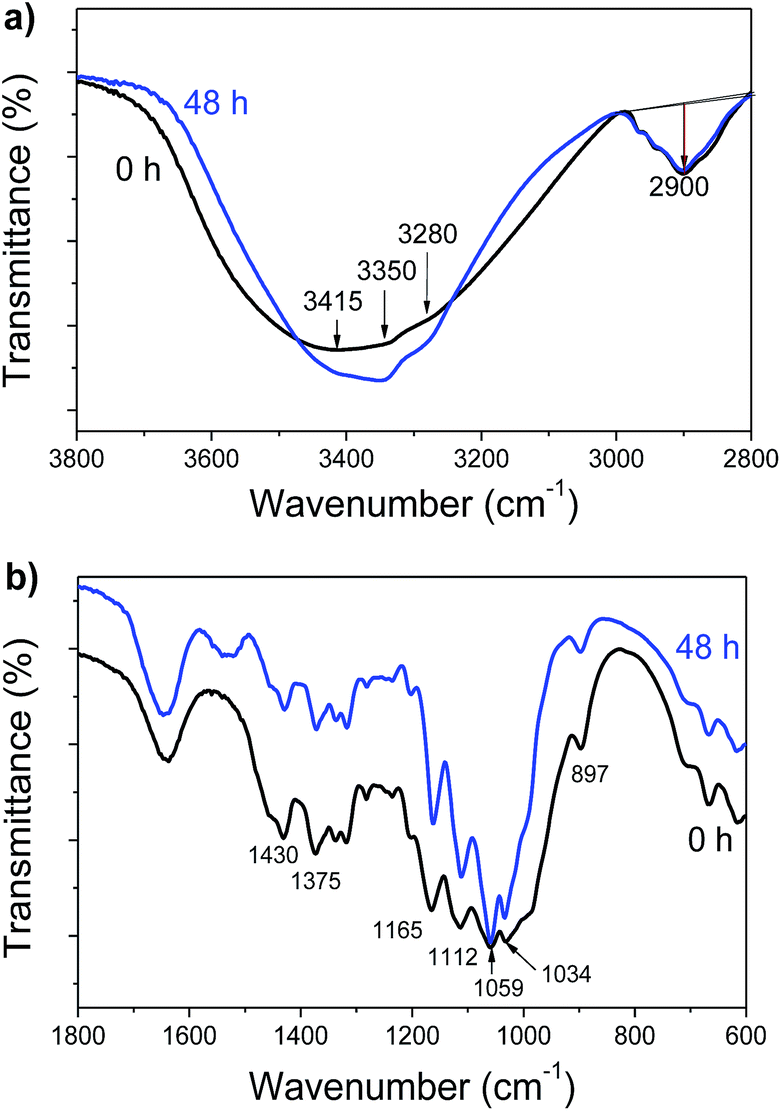 | ||
| Fig. 5 FTIR spectra of Avicel before and after enzymatic hydrolysis in the range of (a) 3800–2800 cm−1 and (b) 1800–600 cm−1. | ||
Fig. 6 shows the FTIR spectra of the corncob samples before and 48 h after the enzymatic hydrolysis. The infrared characteristic peaks and attribution of lignocellulose are shown in Table 3. The band 3420 cm−1, which represented the strong O–H stretching and bending vibrations of the intra- and intermolecular hydrogen bonds in cellulose, shifted to a lower wavelength of 3408 cm−1 and yielded an increase in the intensity after enzymatic hydrolysis. It suggested an increase in the intra- and intermolecular hydrogen bonding strength for the residual corncob. The absorbance ratios, Ar897, Ar1375 and Ar1430, were also used to evaluate the degree of hydrogen bond destruction in corncob.13 As shown in Table 2, the decreased values of Ar897 and Ar1375 indicated an increase in the hydrogen bonding strength derived from the residual corncob, which was in accordance with the results from THz and SEC-MALLS analysis.27 However, the value of Ar1430 in the corncob increased from 0.981 to 0.988, which may be attributed to the other functional groups from lignin or from the residual hemicellulose. It showed the difficulty of measuring hydrogen bonding strength by FTIR technique. In other words, the FTIR results provided some useful information in support of the observation that the THz-TDS technique could be an effective and credible method to evaluate the hydrogen bonding strength within cellulose.
 | ||
| Fig. 6 FTIR spectra of corncob before and after enzymatic hydrolysis in the range of (a) 3800–2800 cm−1 and (b) 1800–600 cm−1. | ||
| Attribution of characteristic peak | Wavenumber (cm−1) | |
|---|---|---|
| Pretreated corncob | Hydrolyzed-corncob | |
| O–H stretching in hydroxyl groups | 3420 | 3408 |
| C–H stretching in methyl and methylene groups | 2920 | 2920 |
| C–H deformations (asymmetry in methyl groups, –CH3– and –CH2–) | 1462 | 1463 |
| Aromatic skeletal vibrations combined with C–H in plane deformations | 1425 | 1423 |
| Aliphatic C–H stretching in methyl and phenol OH | 1373 | 1371 |
| C–H out-of-plane in positions 2 and 6 (S units) | 896 | 896 |
The inter- and intramolecular hydrogen bonds in cellulose were highly related to the crystalline structure. As mentioned above, after enzymatic hydrolysis, the residual cellulosic substrate had an increased hydrogen bonding strength, possibly due to the preferential degradation of cellulose with lower crystallinity and degrees of polymerization (DP). The change in the cellulose crystallinity and DP were interrelated with the hydrogen bonding strength, which was an important factor for understanding the enzymatic mechanism. The THz-TDS analysis demonstrated in this study provided clear evidence for describing the change in the hydrogen bonding strength of cellulose, which was highly consistent with that in FTIR and SEC-MALLS analysis,27 as well as with the results reported in previous studies.31,48
In this study, THz-TDS, as a quick and non-invasive measurement technique, provided a new method for detecting the change in hydrogen bonding strength within cellulosic substrates. Till date, the THz-TDS had been employed to characterize the protein and bimolecular structures. Here, we attempted to use this technique for evaluating hydrogen bonding strength in cellulosic molecules for the first time, to our knowledge. Some key issues, such as the effects of lignin and hemicellulose on the THz spectra, and the quantitative determination of the hydrogen bonding strength, need to be examined in future studies.
Conclusions
In summary, we have demonstrated the terahertz spectroscopy as an efficient technique for directly evaluating the hydrogen bonding strength within cellulose. The increased THz adsorption and refractive index provided a clear signal of an increase in the hydrogen bonding strength in the residual Avicel and corncob after enzymatic hydrolysis. The THz results were further compared with the XRD and FTIR analysis, as well as with the SEC-MALLS results reported in our previous study. The THz method demonstrated in this study opens a new avenue for evaluating the change in the hydrogen bonding strength in cellulose and thus provides some molecular information for understanding the mechanism of cellulose degradation.Acknowledgements
We acknowledge the financial supports received from the National Natural Science Foundation of China (no. 21276192), the Ministry of Science and Technology of China (no. 2012BAD29B05 and 2013AA102204), Open Funding Project of the State Key Laboratory of Chemical Engineering (no. SKL-ChE-11B01), and the Ministry of Education (no. NCET-11-0372, 20110032130004, and B06006).Notes and references
- R. Huang, R. Su, W. Qi and Z. He, BioEnergy Res., 2011, 4, 225–245 CrossRef.
- H. Jørgensen, J. B. Kristensen and C. Felby, Biofuels, Bioprod. Biorefin., 2007, 1, 119–134 CrossRef.
- H. Chen and W. Qiu, Biotechnol. Adv., 2010, 28, 556–562 CrossRef CAS PubMed.
- S. P. S. Chundawat, G. Bellesia, N. Uppugundla, L. D. Sousa, D. H. Gao, A. M. Cheh, U. P. Agarwal, C. M. Bianchetti, G. N. Phillips, P. Langan, V. Balan, S. Gnanakaran and B. E. Dale, J. Am. Chem. Soc., 2011, 133, 11163–11174 CrossRef CAS PubMed.
- L. T. Fan, Y.-H. Lee and D. H. Beardmore, Biotechnol. Bioeng., 1980, 22, 177–199 CrossRef CAS.
- B. Yang and C. E. Wyman, Biotechnol. Bioeng., 2004, 86, 88–98 CrossRef CAS PubMed.
- M. Monrroy, I. Ortega, M. Ramírez, J. Baeza and J. Freer, Enzyme Microb. Technol., 2011, 49, 472–477 CrossRef CAS PubMed.
- C. M. Mooney, S. D. Mansfield and J. N. Saddler, Biotechnol. Prog., 1999, 15, 804–816 CrossRef PubMed.
- S. Park, J. O. Baker, M. E. Himmel, P. A. Parilla and D. K. Johnson, Biotechnol. Biofuels, 2010, 3, 10 CrossRef PubMed.
- R. Huang, R. Su, W. Qi and Z. He, Biotechnol. Prog., 2010, 26, 384–392 CrossRef CAS PubMed.
- B. Medronho, A. Romano, M. G. Miguel, L. Stigsson and B. Lindman, Cellulose, 2012, 19, 581–587 CrossRef CAS.
- Y. Maréchal and H. Chanzy, J. Mol. Struct., 2000, 523, 183–196 CrossRef.
- K. Kamide, K. Okajima and K. Kowsaka, Polym. J., 1992, 24, 71–86 CrossRef CAS.
- S. Y. Oh, D. I. Yoo, Y. Shin and G. Seo, Carbohydr. Res., 2005, 340, 417–428 CrossRef CAS PubMed.
- X. Colom and F. Carrillo, Eur. Polym. J., 2002, 38, 2225–2230 CrossRef CAS.
- R. Pönni, E. Kontturi and T. Vuorinen, Carbohydr. Polym., 2013, 93, 424–429 CrossRef PubMed.
- M. Suchy, E. Kontturi and T. Vuorinen, Biomacromolecules, 2010, 11, 2161–2168 CrossRef CAS PubMed.
- M. Zhang, W. Qi, R. Liu, R. Su, S. Wu and Z. He, Biomass Bioenergy, 2010, 34, 525–532 CrossRef CAS PubMed.
- M. Wada, L. Heux, Y. Nishiyama and P. Langan, Cellulose, 2009, 16, 943–957 CrossRef CAS.
- M. Hangyo, M. Tani and T. Nagashima, Int. J. Infrared Millimeter Waves, 2005, 26, 1661–1690 CrossRef CAS PubMed.
- C. Yan, B. Yang and Z. C. Yu, Analyst, 2014, 139, 1967–1972 RSC.
- A. Arora, T. Q. Luong, M. Kruger, Y. J. Kim, C. H. Nam, A. Manz and M. Havenith, Analyst, 2012, 137, 575–579 RSC.
- A. Redo-Sanchez, G. Salvatella, R. Galceran, E. Roldos, J. A. Garcia-Reguero, M. Castellari and J. Tejada, Analyst, 2011, 136, 1733–1738 RSC.
- Y. C. Shen, P. C. Upadhya, E. H. Linfield and A. G. Davies, Appl. Phys. Lett., 2003, 82, 2350 CrossRef CAS PubMed.
- R. Liu, M. He, R. Su, Y. Yu, W. Qi and Z. He, Biochem. Biophys. Res. Commun., 2010, 391, 862–867 CrossRef CAS PubMed.
- T. Bardon, R. K. May, P. F. Taday and M. Strlic, Analyst, 2013, 138, 4859–4869 RSC.
- R. Y. Du, R. L. Huang, R. X. Su, M. J. Zhang, M. F. Wang, J. F. Yang, W. Qi and Z. M. He, RSC Adv., 2013, 3, 1871–1877 RSC.
- M. Zhang, R. Su, W. Qi, R. Du and Z. He, Chin. J. Chem. Eng., 2011, 19, 773–778 CrossRef CAS.
- L. Segal, J. J. Creely, A. E. Martin and C. M. Conrad, Text. Res. J., 1959, 29, 786–794 CrossRef CAS PubMed.
- M. Hall, P. Bansal, J. H. Lee, M. J. Realff and A. S. Bommarius, FEBS J., 2010, 277, 1571–1582 CrossRef CAS PubMed.
- L. Wang, Y. Zhang, P. Gao, D. Shi, H. Liu and H. Gao, Biotechnol. Bioeng., 2006, 93, 443–456 CrossRef CAS PubMed.
- S. Yang, J. Li, Z. Zheng and Z. Meng, Int. Biodeterior. Biodegrad., 2009, 63, 569–575 CrossRef CAS PubMed.
- L. Hao, R. Wang, L. Zhang, K. Fang, Y. Men, Z. Qi, P. Jiao, J. Tian and J. Liu, Cellulose, 2013, 21, 777–789 CrossRef PubMed.
- H. Kargarzadeh, I. Ahmad, I. Abdullah, A. Dufresne, S. Y. Zainudin and R. M. Sheltami, Cellulose, 2012, 19, 855–866 CrossRef CAS.
- H. Zhao, J. Kwak, Z. Conradzhang, H. Brown, B. Arey and J. Holladay, Carbohydr. Polym., 2007, 68, 235–241 CrossRef CAS PubMed.
- S. Kim and M. T. Holtzapple, Bioresour. Technol., 2006, 97, 583–591 CrossRef CAS PubMed.
- L. Zhu, J. P. O'Dwyer, V. S. Chang, C. B. Granda and M. T. Holtzapple, Bioresour. Technol., 2008, 99, 3817–3828 CrossRef CAS PubMed.
- H. Yoneyama, M. Yamashita, S. Kasai, K. Kawase, R. Ueno, H. Ito and T. Ouchi, Phys. Med. Biol., 2008, 53, 3543 CrossRef CAS PubMed.
- J. Xu, K. W. Plaxco and S. J. Allen, Protein Sci., 2006, 15, 1175–1181 CrossRef CAS PubMed.
- J. Y. Chen, J. R. Knab, J. Cerne and A. G. Markelz, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 72, 040901 CrossRef.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- R. Huang, S. Wu, A. Li and Z. Li, J. Mater. Chem. A, 2014, 2, 1672–1676 CAS.
- Y. Lu, G. Zhang, M. Feng, Y. Zhang, M. Yang and D. Shen, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 2313–2321 CrossRef CAS.
- M. Walther, B. M. Fischer and P. Uhd Jepsen, Chem. Phys., 2003, 288, 261–268 CrossRef CAS.
- S. Y. Oh, D. I. Yoo, Y. Shin, H. C. Kim, H. Y. Kim, Y. S. Chung, W. H. Park and J. H. Youk, Carbohydr. Res., 2005, 340, 2376–2391 CrossRef CAS PubMed.
- T. Kondo and C. Sawatari, Polymer, 1996, 37, 393–399 CrossRef CAS.
- A. Bazargan, J. Tan, C. W. Hui and G. McKay, Cellulose, 2014, 21, 1679–1688 CrossRef CAS PubMed.
- Z. Liao, Z. Huang, H. Hu, Y. Zhang and Y. Tan, Bioresour. Technol., 2011, 102, 7953–7958 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra08314h |
| This journal is © The Royal Society of Chemistry 2014 |