
Novel magnetic multi-template molecularly imprinted polymers for specific separation and determination of three endocrine disrupting compounds simultaneously in environmental water samples†
Abstract
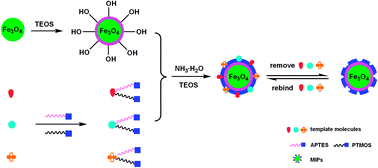
In order to improve the practical applied value of molecularly imprinted polymers, a novel concept of multiple templates was introduced to prepare an original type of magnetic molecularly imprinted polymers. The magnetic multi-template molecularly imprinted polymers were obtained by selecting silica-coated magnetic nanoparticles as supporters, three endocrine disrupting compounds (17β-estradiol (E2), estriol (E3), and diethylstilbestrol (DES)) as the multi-template, and two kinds of silane coupling agents (3-aminopropyltriethoxysilane (APTES) and phenyltrimethoxysilane (PTMOS)) as bifunctional monomers for simultaneously specific recognition of E2, E3, and DES. The as-synthesized polymers possessed homogeneous imprinting shells, stable crystalline phase, and super-paramagnetic properties. Meanwhile, the imprinted nanomaterials displayed not only extraordinarily fast kinetics, but also satisfactory adsorption capacity, as well as favorable selectivity. More importantly, the prepared polymers exhibited a similar recognition performance to a physical mixture of three single-template polymers, but the synthetic procedure of the former was simplified with significant reduction in both preparation time and solvent consumption. In addition, the imprinted nanoparticles were applied as a specific adsorbent coupled with HPLC-UV for rapid isolation and determination of E2, E3, and DES simultaneously. The limits of detection (LODs) of proposed method for three target estrogens of E2, E3, and DES were 0.27, 0.19, and 0.08 ng mL−1, respectively, which were lower than that obtained by some other sample pretreatment methods followed by HPLC-UV analysis. Furthermore, the developed method was successfully applied for detection of multiple aimed estrogens in environmental water samples with satisfactory recoveries in the range of 92.3–98.6%.
 Please wait while we load your content...
Please wait while we load your content...

