
Photocatalytically green synthesis of H2O2 using 2-ethyl-9,10-anthraquinone as an electron condenser†
Dandan Zhang,
Gangqiang Xu,
Tao Chen and
Feng Chen*
Key Laboratory for Advanced Materials, Institute of Fine Chemicals, East China University of Science and Technology, 130 Meilong Road, Shanghai, 200237, PR China. E-mail: fengchen@ecust.edu.cn
First published on 10th October 2014
Abstract
A high level (∼9 mM) of hydrogen peroxide is photocatalytically synthesized by using O2 as an oxidant and 2-ethyl-9,10-anthraquinone (EAQ) as an electron condenser. The photo-catalytic efficiency of the EAQ-assisted H2O2 production is ca. 10-fold higher than that of H2 generation with Pt/P25.
Hydrogen peroxide (H2O2) is a clean oxidant that emits only water as a byproduct and is widely used in industries for organic synthesis,1 pulp bleaching, waste-water treatment, and as a disinfectant.2 Traditionally, H2O2 is produced with an anthraquinone oxidation (AO) process which combines H2 with O2 through a multistep route via an anthraquinone/anthrahydroquinone cycle. Recently, from the viewpoint of green and sustainable chemistry, direct synthesis of H2O2 from H2 and O2 has attracted a lot of attention.3 However, the potentially explosive nature of H2/O2 gas mixture brings a serious risk to the operation process; further, highly pure H2 gas is indispensable which requires significant energy costs in both the AO process and the H2/O2 direct synthesis process. To circumvent the above disadvantages, photocatalytic attempts have begun to emerge nowadays.4 By using methanol/ethanol as a sacrificial electron donor, direct synthesis of H2O2 is reported to be available.5
However, direct photocatalytic generation of H2O2 from methanol is not an easy task due to the undesirable rapid reverse reactions5c as well as the over-reduction of surface hydroperoxo species.6 As we know, the photocatalytic production of H2O2 is a multistep process, which is briefly shown below.5c,7,8
| Ti3+(e−) + O2 → Ti4+–O2˙− | (1) |
| Ti3+(e−)–O2˙− + H+ → Ti4+–−O2H | (2) |
| Ti4+–−O2H + H+ → Ti4+ + H2O2 | (3) |
The photoelectron on the TiO2 surface, Ti3+(e−), reduces the adsorbed O2 and produces a superoxo radical.5a Then the superoxo radical is transformed to a hydroperoxo specie via further reduction, and protonated to produce H2O2.7,8 Unfortunately, the hydroperoxo species are easily decomposed by further reduction with e− to produce hydroxyl radical,8 which has been frequently verified with Electron Paramagnetic Resonance (EPR) technique.8,9
| Ti3+(e−)–−O2H + H+ → Ti4+–˙OH + HO− | (4) |
Also, the back reaction of the hydroperoxo specie with photo-holes is highly possible.6
| Ti4+–−O2H + h+ → Ti4+–O2H ↔ Ti4+–O2˙− + H+ | (5) |
So far, H2O2-assisted photocatalysis is inevitable,10 which limits the H2O2 concentration (from tens of μM to several mM) in most reported systems for directly photocatalytic H2O2 production. Hereinafter, we introduce an experimental attempt to photocatalytically produce H2O2 of high levels by innovatively employing 2-ethyl-9,10-anthraquinone (EAQ) as an electron condenser. The photogenerated electrons are trapped by EAQ during the photocatalytic process by forming 2-ethyl-9,10-anthrahydroquinone (H2EAQ), which subsequently induces reduction of O2 molecules into H2O2 by simple air bubbling in dark (eqn (6), Fig. S1†).
| H2EAQ + O2 → EAQ + H2O2 | (6) |
Pt nanoclusters deposited P25 TiO2 (Pt/P25, with a Pt percent of 0.4 wt%) was employed as the photocatalyst, which was prepared with a typical photocatalytic reduction method11 and had a light gray color. Pt/P25 contains just similar TiO2 lattice phase composition with that of P25 (rutile/anatase: 20/80, Fig. S2†), of which Pt nanoclusters of ca. 2 nm are well deposited on the TiO2 grains (20–30 nm, Fig. S3†). Alternatively, some other TiO2 composites, such as Ag/TiO212a and MoS2/TiO2,12b can also be used as the photocatalyst in this work.
The photocatalytic reaction was carried out by using methanol (100 mL) as the solvent and the hole scavenger, EAQ as the electron condenser, and N2 bubbling to keep the system anaerobic. The dosage of Pt/P25 was 0.2 g. Pt/P25 has significant photoabsorption at wavelength less than 406 nm (Fig. S4†), while EAQ extends its photoabsorption to 471 nm (Fig. S4†). Hence, the reaction solution was irradiated under either direct irradiation (300 W Xe lamp, PLS-SXE300) or visible irradiation (with an optical filter, λ > 420 nm). Fig. 1A shows the photocatalytic generation of H2O2 and the corresponding control reactions. H2O2 is kept absent throughout the reaction under visible irradiation with photocatalyst or under direct light irradiation without the Pt/P25 photocatalyst, but generated smoothly under direct irradiation with photocatalyst. As EAQ absorbs both UV and visible light while TiO2 can only be aroused with UV light, it suggests that the H2O2 is generated via a photocatalysis-related process. Briefly, photogenerated electrons in the Pt/P25 photocatalyst reduce the EAQ molecules to give H2EAQ, while the holes at the valence band of Pt/P25 are captured by hole scavenger, methanol. When H2EAQ solution is air bubbled, the reaction of oxygen with H2EAQ leads to the formation of H2O2. Notably, the H2O2 production rate is ca. 10-fold than that of H2 with Pt/P25 according to their initial slopes as shown in Fig. 1A.
 | ||
| Fig. 1 (A) Photocatalytic generation of H2O2 under (a) direct irradiation with photocatalyst, (b) visible irradiation with photocatalyst, (c) direct irradiation without photocatalyst, and (d) photocatalytic generation of H2 under direct irradiation. (B) H2O2 generation with EAQ dosages of (a) 1.0 g L−1, (b) 2.0 g L−1, (c) 3.0 g L−1, (d) 4.0 g L−1, (e) 6.0 g L−1 with the Pt/P25 photocatalyst. | ||
Fig. 1B shows the generating profiles of H2O2 with various EAQ dosages in this photocatalytic system. The H2O2 is initially produced efficiently in all cases; however, at the EAQ dosage of 1.0 g L−1, the H2O2 production gets slower and reaches its maximum at a reaction time of ca. 1.5 h. The measured and the fitted maximal produced H2O2 concentrations are 2.6 mM and 2.5 mM, respectively. Further prolonging the irradiation time leads to a slow decrease in H2O2 concentration. The exhaustion of EAQ would be the main constraint that limits the maximal H2O2 level in this reaction. Meanwhile, two possible reasons can also be responsible for the adverse H2O2 concentration decrease, which we observed in Fig. 1B: one is the back-reaction of H2EAQ with the photogenerated holes, which slowly becomes serious at higher H2EAQ concentration. The other is the chemical deterioration of EAQ; as we know, the chemical structure change of EAQ, especially on its anthracene ring, would alter the redox potential of EAQ/H2EAQ and disable the H2O2 production from O2 via H2EAQ.
Although a H2O2 concentration of 2.5 mM is quite high among the literature reported H2O2 levels with the directly photocatalytic synthesis,4 optimizing the reaction parameters to get a better production is preferred. Increasing the EAQ dosage to 2.0 g L−1 changes much on the H2O2 generation. As shown in Fig. 1B, the H2O2 concentration further increases and reaches its maximum (5.3 mM) at an irradiation time of 3.9 h, which is ca. 2.1-fold than that with EAQ dosage of 1.0 g L−1. As shown in Table 1 (also Table S1†), the maximal H2O2 concentrations become higher at higher EAQ dosages; a maximal H2O2 concentration of 9.1 mM is produced with EAQ dosage of 4.0 g L−1 at a reaction time of 8.2 h, which should be ascribed to that more concentrated EAQ benefits the photoelectron scavenge and thus favors the formation of H2EAQ. However, as the light absorption region of TiO2 mostly overlaps with that of EAQ, increasing the EAQ dosage seems adverse to the initial generation rate of H2O2. EAQ of high concentrations would significantly compete with Pt/P25 on photon absorption and thus hinder some the photoexcitation of Pt/P25 photocatalyst. Nevertheless, a high initial EAQ dosage would enhance the reachable H2EAQ level under irradiation; i.e., the generated amounts of H2O2 with higher EAQ dosages would slowly catch up with and even surpass those with lower EAQ dosages as shown in Fig. 1B. However, much longer irradiation times are required for reaching the maximal H2EAQ levels with the higher EAQ dosages.
The side reactions during the EAQ-assisted photocatalytic H2O2 production are undesired and adverse in this work, just as those in the industrial AO process. Therefore, the intermediates and the products of the reaction solution were monitored with HPLC (Fig. S5†) and LC-MS (Fig. S6 and S12†) techniques. Table 2 lists all the substances that were detected out in the solution. Correspondingly, a schematically photo catalytic reaction of EAQ is proposed in Fig. 2. H2EAQ is obtained from EAQ via two successive single-electron reduction processes. However, an inverse oxidation is also possible to the semi-hydroanthraquinone intermediate, which leads to the formation of oxo-H2EAQ1 and oxo-H2EAQ2 intermediates. Due to the strong reducibility of H2EAQ, oxo-H2EAQ1 and oxo-H2EAQ2, they would quickly react with oxygen molecules to produce H2O2 and transform themselves back to EAQ, oxo-EAQ1 and oxo-EAQ2, respectively. A prolonged photocatalytic process induces a deep hydrogenation of H2EAQ, which results the formation of 2-ethyl-9-anthranol, 2-ethyl-10-anthranol, 2-ethyl-10-H-9-anthranone (EAN1) and 2-ethyl-9-H-10-anthranone (EAN2). These four deep hydrogenation products are not strange interlopers. They are unavoidable byproducts in AO process2 and considered unavailable to produce H2O2. However, the last substance (isomer of EAQ) identified from the reaction solution suggests the formation of another EAQ (which differs only in the position of quinone groups to the original EAQ molecule), which is possibly the oxidation product from EAN1 and EAN2.
| Time (min) | Formula | Substance | Structure | Meas. m/z | Pred. Mw |
|---|---|---|---|---|---|
| 11.24 | C16H12O3 | Oxo-EAQ1 |  |
253.120 | 252.122 |
| 11.52 | C16H12O3 | Oxo-EAQ2 |  |
253.120 | 252.122 |
| 14.98 | C16H14O | 2-Ethyl-9-anthranol |  |
223.110 | 222.111 |
| 15.27 | C16H14O | 2-Ethyl-10-anthranol |  |
223.109 | 222.111 |
| 17.32 | C16H14O | EAN1 |  |
223.108 | 222.111 |
| 17.79 | C16H14O | EAN2 | 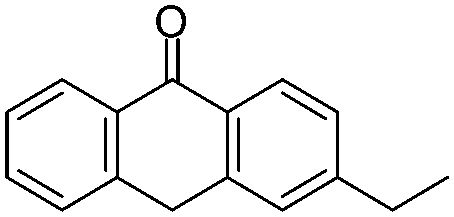 |
223.108 | 222.111 |
| 18.83 | C16H12O2 | EAQ |  |
— | — |
| 20.41 | C16H12O2 | EAQ isomer | — | 237.089 | 236.091 |
 | ||
| Fig. 2 Proposed photocatalytic reduction reaction pathways for EAQ. | ||
Conclusions
In summary, a novel way is introduced to photocatalytically produce H2O2 of high levels by employing EAQ as an electron condenser. A concentration of H2O2 with 9.1 mM was obtained with 4.0 g L−1 EAQ for 8.2 h irradiation, which is advantageous comparing with the directly photocatalytic route for H2O2 production (from tens of μM to several mM). The enhanced H2O2 formation is due to the electron condensing with EAQ and the “off-site” H2O2 generation strategy, which circumvents the adverse over-reduction or back-reaction of hydroperoxo species (the precursor of H2O2) during the photocatalytic process. It suggests a potential candidate route toward the green and safe H2O2 synthesis without producing highly pure H2 in advance.Acknowledgements
This work was supported by the National Nature Science Foundations of China (21177039) and the Innovation Program of Shanghai Municipal Education Commission (13ZZ042).Notes and references
- K. Sato, M. Aoki and R. Noyori, Science, 1998, 281, 1646 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962 CrossRef CAS PubMed.
- (a) J. H. Lunsford, J. Catal., 2003, 216, 455 CrossRef CAS; (b) J. K. Edwards, B. Solsona, E. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037 CrossRef CAS PubMed; (c) G. Blanco-Brieva, E. Cano-Serrano, J. M. Campos-Martin and J. L. Fierro, Chem. Commun., 2004, 1184 RSC; (d) P. Landon, P. J. Collier, A. J. Papworth, C. J. Kiely and G. J. Hutchings, Chem. Commun., 2002, 2058 RSC; (e) S. Melada, R. Rioda, F. Menegazzo, F. Pinna and G. Strukul, J. Catal., 2006, 239, 422 CrossRef CAS PubMed; (f) V. R. Choudhary, A. G. Gaikwad and S. D. Sansare, Angew. Chem., Int. Ed., 2001, 40, 1776 CrossRef CAS.
- (a) H. Goto, Y. Hanada, T. Ohno and M. Matsumura, J. Catal., 2004, 225, 223 CrossRef CAS PubMed; (b) Y. Yamada, A. Nomura, T. Miyahigashi and S. Fukuzumi, Chem. Commun., 2012, 48, 8329 RSC; (c) M. Fukushima, K. Tatsumi, S. Tanaka and H. Nakamura, Environ. Sci. Technol., 1998, 32, 3948 CrossRef CAS.
- (a) M. Teranishi, S. Naya and H. Tada, J. Am. Chem. Soc., 2010, 132, 7850 CrossRef CAS PubMed; (b) D. Tsukamoto, A. Shiro, Y. Shiraishi, Y. Sugano, S. Ichikawa, S. Tanaka and T. Hirai, ACS Catal., 2012, 2, 599 CrossRef CAS; (c) T. Hirakawa and Y. Nosaka, J. Phys. Chem. C, 2008, 112, 15818 CrossRef CAS; (d) V. Maurino, C. Minero, G. Mariella and E. Pelizzetti, Chem. Commun., 2005, 2627 RSC.
- R. Nakamura and Y. Nakato, J. Am. Chem. Soc., 2004, 126, 1290 CrossRef CAS PubMed.
- T. Hirakawa, T. Daimon, M. Kitazawa, N. Ohguri, C. Koga, N. Negishi, S. Matsuzawa and Y. Nosaka, J. Photochem. Photobiol., A, 2007, 190, 58 CrossRef CAS PubMed.
- R. Nakamura, A. Imanishi, K. Murakoshi and Y. Nakato, J. Am. Chem. Soc., 2003, 125, 7443 CrossRef CAS PubMed.
- (a) X. Li, C. Chen and J. Zhao, Langmuir, 2001, 17, 4118 CrossRef CAS; (b) F. Herrera, J. Kiwi, A. Lopez and V. Nadtochenko, Environ. Sci. Technol., 1999, 33, 3145 CrossRef CAS.
- R. Cai, Y. Kubota and A. Fujishima, J. Catal., 2003, 219, 214 CrossRef CAS.
- P. Chowdhury, H. Gomaa and A. K. Ray, Int. J. Hydrogen Energy, 2011, 36, 13442 CrossRef CAS PubMed.
- (a) H. Li, W. Lu, J. Tian, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, Chem.–Eur. J., 2012, 18, 8508 CrossRef CAS PubMed; (b) Q. Liu, Z. Pu, A. M. Asiri, A. H. Qusti, A. O. Al-Youbi and X. Sun, J. Nanopart. Res., 2013, 15, 2057 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterizations of the Pt/P25 catalyst, photocatalytic H2O2 synthesis, determination of H2O2, hydrogen evolution, by-products analysis with HPLC and LC-MS, mechanism of H2O2 generation, reduction kinetics of EAQ and the corresponding reaction rate constants. See DOI: 10.1039/c4ra09702e |
| This journal is © The Royal Society of Chemistry 2014 |