
Safety evaluation of graphene oxide-based magnetic nanocomposites as MRI contrast agents and drug delivery vehicles†
Abstract
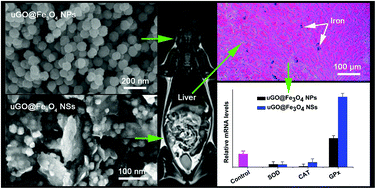
A variety of graphene oxide-based magnetic nanocomposites have been developed based on the purpose for biomedical applications. However, their safety is still unclear in vivo. Here, two nanocomposites of ultrafine graphene oxide-iron oxide nanoparticles and nanosheets (uGO@Fe3O4 NPs and NSs) were prepared. The samples were used for evaluating the morphology, dosage, time-dependent effects on various biochemical parameters of clinical significances at various levels: organ, cell, and molecule in a female Wistar mouse model and hepatic cell lines. At the organ level, the explorations of biodistribution, MRI, and histopathology consistently showed that the uGO@Fe3O4 NPs and NSs were primarily trapped in the liver and spleen. The heart, liver, spleen, and kidney were damaged based on histopathological observation and the obvious fluctuation of blood biochemical indicators. At the cellular level, the studies of blood compatibility and cytotoxicity demonstrated that the uGO@Fe3O4 NSs possessed a higher toxicity than uGO@Fe3O4 NPs, and resulted in about 47% (HepG2) and 48% (L02) loss of cell viability at their highest concentration (400 μg Fe per mL). At the molecular level, uGO@Fe3O4 NPs and NSs suppressed mRNA expression levels of SOD and upregulated mRNA expression levels of GPx, while the effect on CAT gene expression was mixed, suggesting the significant fluctuation in uGO@Fe3O4 nanocomposites-induced oxidative stress in mice. The assessments indicated that the uGO@Fe3O4 NPs and NSs have no acute fatal toxicity, but do have some toxicity.
 Please wait while we load your content...
Please wait while we load your content...

