DOI:
10.1039/C4RA09574J
(Paper)
RSC Adv., 2014,
4, 56102-56111
Palladium(II) complexes bearing the 1,2,3-triazole based organosulfur/ selenium ligand: synthesis, structure and applications in Heck and Suzuki–Miyaura coupling as a catalyst via palladium nanoparticles†
Received
1st September 2014
, Accepted 20th October 2014
First published on 21st October 2014
Abstract
Air and moisture insensitive palladium complexes, [Pd(L)Cl2] (1/2), in which L = 1-benzyl-4-phenylthiomethyl or 1-benzyl-4-phenylselenomethyl-1H-1,2,3-triazole (L1 or L2) catalyze Heck (HC) and Suzuki–Miyaura coupling (SMC) reactions between a series of aryl bromides including deactivated bromides and n-butyl acrylate and phenylboronic acid, respectively. The optimal catalytic loading was found to be in the order of 0.01 mol%. HRTEM, TGA and EDX data indicated that 3–11 nm nanoparticles (NPs) composed of palladium and sulfur or selenium and protected with L or its fragment, were formed during the catalyzed reaction. The isolated NPs displayed catalytic activity and appeared to have a role in the catalysis. A two-phase test indicated that both homogeneous and heterogeneous catalysis took place. The complexes 1 and 2 were synthesized by the reactions of L1 and L2 respectively with [(MeCN)2PdCl2]. Their single crystal X-ray diffraction indicated that the geometry adopted by ligands around Pd in both complexes is distorted square planar with Pd–S and Pd–Se bond lengths of 2.2727(14) and 2.3693(8) Å, respectively. DFT calculation gave bond lengths and angles in keeping with the experimental values. The DFT calculated HOMO–LUMO energy difference is lower for 1 than for 2 in accordance with the observed higher catalytic activity of 1.
Introduction
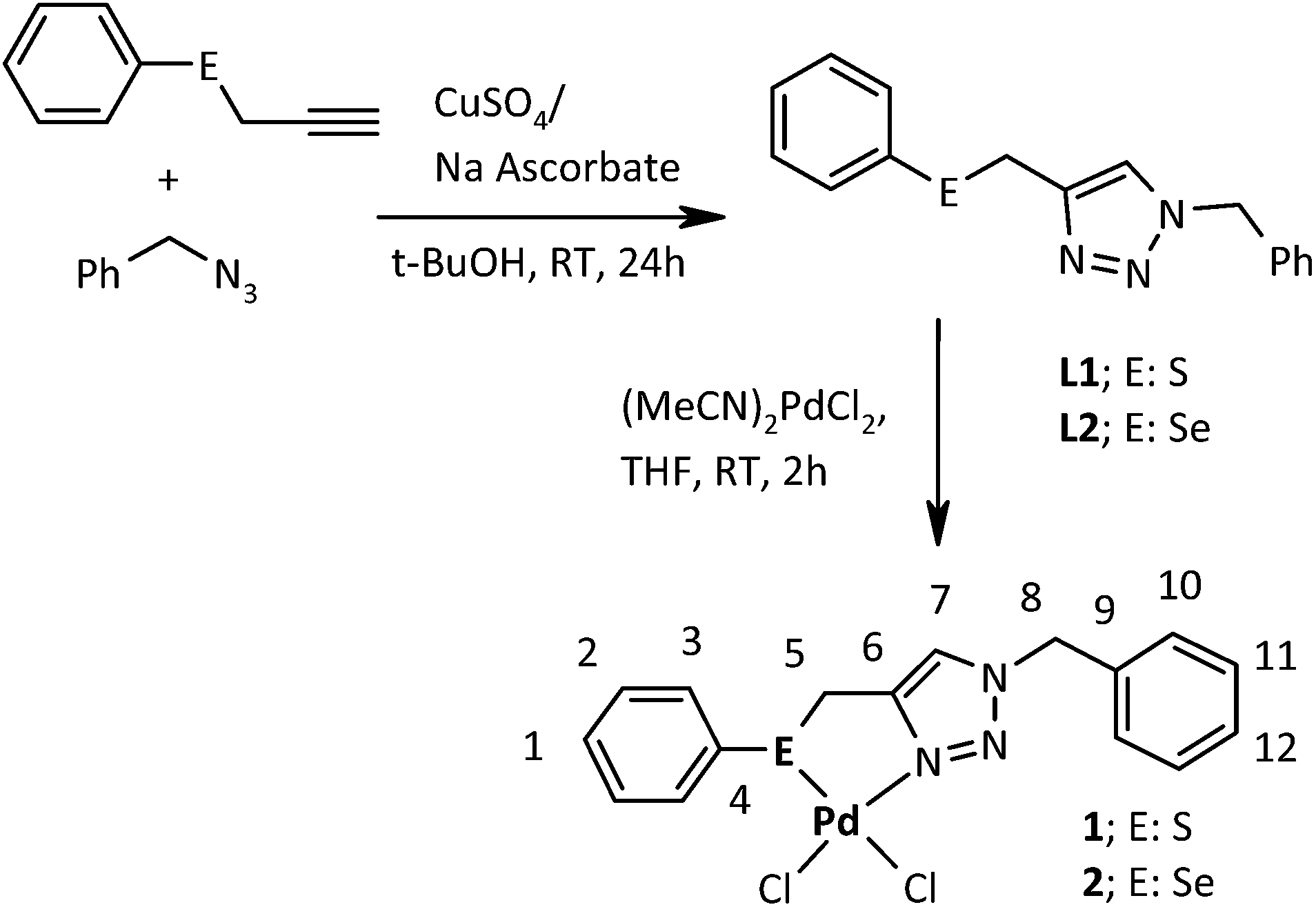
Carbon–carbon bond forming reactions have emerged as exceedingly important methodologies for the preparation of complex organic molecules.1 Of the several commonly used C–C coupling reactions, such as Heck, Suzuki, Sonogashira and Stille, the two most important ones are the Heck2–4 and Suzuki coupling.5,6 These two are fascinating from a catalysis perspective. Virtually, all forms of palladium including molecular complexes,7–9 nanoparticles9–12 and salts13,14 have been used as catalyst/pre-catalysts for activation of coupling of aryl iodides and some aryl bromides. Specifically designed catalysts are required for activation of bulky or electronically unactivated substrates e.g. aryl chlorides.15–18 The catalysts for Heck and Suzuki coupling include palladium complexes of several phosphorus,15,16,18–24 carbene,15,17 amino acids,25a,b imine25c and related ligands. However, many of phosphorous ligated Pd complexes are prone to oxidation and sensitive to moisture. Thus argon or nitrogen atmosphere22,23 is required during catalysis to protect catalyst from deactivation. In view of moisture and air sensitivity of these complexes, a number of Pd(II) species bearing chalcogen (S, Se or Te) ligands,26–28 have been explored as possible alternatives to catalyse various chemical transformations29,30 including C–C coupling reactions.31–48 The catalytic activity of some of these complexes (thermally stable and resistant to aerial oxidation) not only rivals but vastly outperforms that of the corresponding phosphorus analogues.47 Some catalysts of this class have also been reported efficient for coupling32,34 of aryl chlorides, which are the cheapest and most readily available among the aryl halides. Suzuki coupling reactions catalyzed with Pd–chalcogen ligand complexes are reported to proceed via in situ formation of nanoparticles or discrete palladium(0) and the role of ligand appears to be limited to stabilize Pd(0) species or affect dispersion, size and chemical nature of NPs.32,34 In favour of such in situ generation and participation of palladium39,43 and palladium chalcogenide35,44,46 nanoparticles in Suzuki coupling there are reasonable evidences.38 However, there are very few reports in case of Heck coupling where formation and involvement of palladium nano-phases have been described.32,33 Further these reports are without sufficient experimental support.32 Herein we report that formation of NPs is almost parallel for Heck and Suzuki coupling, when palladium(II) complexes 1 and 2 of ligands L1 and L2 (Scheme 1) were used as catalysts. These ligands are 1,2,3-triazole based and in the recent past, applications of such organochalcogen ligands in designing catalysts for transfer hydrogenation and oxidation have been reported by our group.29,30 The synthesis of palladium complexes of a (P, C, S) pincer ligands having 1,2,3-triazole skeleton and their applications in Heck coupling are known.49 The presence of 1,2,3-triazole unit in a ligand offers an interesting alternative to the traditional phenyl based framework and has been found to render interesting variation in catalytic activities of metal complexes of such ligands, due to potential of this unit to act as a N donor through two types of nitrogen atoms: viz., N(2) and N(3).29 The ligands L1 and L2 (Scheme 1) have been designed by click reaction (1,3-dipolar cycloaddition) and treatment of [(MeCN)2PdCl2] with them results in complexes 1 and 2 respectively. The major advantages of these catalysts include high activity in the absence of any additive and easy synthesis including those of corresponding ligands. DFT calculations support the difference in activities of 1 and 2.
 |
| | Scheme 1 Methodology for syntheses of ligands and their complexes. | |
Results and discussion
The syntheses of L1, L2 and their complexes have been summarized in Scheme 1. Earlier reported detailed procedures29,30 were used for the preparation of L1 and L2. These air and moisture insensitive ligands were found to show good solubility in CHCl3, CH2Cl2, CH3OH and CH3CN. Complexes 1 and 2 as yellow solids were synthesized by reacting [(MeCN)2PdCl2] with L1 and L2 respectively in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio, in tetrahydrofuran at room temperature. The yield was 90 and 85% for 1 and 2 respectively. The 1 and 2 can be stored in aerobic conditions, as no degradation was indicated by NMR even after six months. The two air and moisture insensitive complexes are moderately soluble in CHCl3, CH2Cl2 and CH3OH, but have good solubility in CH3CN and DMSO. The L1 and L2 and their Pd complexes, 1 and 2, were characterized with 1H, 13C{1H} and 77Se{1H} NMR, HR-MS and IR spectra. The 1H, 13C{1H}, and 77Se{1H} NMR and mass spectra of L1 and L2 were found matching with those reported earlier.29 These spectra for complexes 1 and 2 are given in the ESI (Fig. S1–S7†).
:1 molar ratio, in tetrahydrofuran at room temperature. The yield was 90 and 85% for 1 and 2 respectively. The 1 and 2 can be stored in aerobic conditions, as no degradation was indicated by NMR even after six months. The two air and moisture insensitive complexes are moderately soluble in CHCl3, CH2Cl2 and CH3OH, but have good solubility in CH3CN and DMSO. The L1 and L2 and their Pd complexes, 1 and 2, were characterized with 1H, 13C{1H} and 77Se{1H} NMR, HR-MS and IR spectra. The 1H, 13C{1H}, and 77Se{1H} NMR and mass spectra of L1 and L2 were found matching with those reported earlier.29 These spectra for complexes 1 and 2 are given in the ESI (Fig. S1–S7†).
NMR spectra
The 1H, 13C{1H} and 77Se{1H} NMR spectra of complexes 1 and 2 were found in agreement with their molecular structures depicted in Scheme 1. The signal in the 77Se{1H} NMR spectrum of 2 appears shifted to a higher frequencies by ∼95 ppm with respect to that of free L2, observed at 364 ppm. Probably it arises due to coordination of L2 with Pd through Se. In 1H and 13C{1H} NMR spectra of 1 and 2, signals of protons and carbon atoms appear at higher frequencies relative to those of free ligands. In 13C{1H} NMR spectra magnitude of high frequency shift for C5 (SCH2) was found to be 6.2 ppm in case of sulfur ligand and 7.6 ppm for selenium analogue. In 1H NMR spectra signals of H5 protons attached to C5 also appear at a higher frequency (≤0.44 ppm) relative to those of free ligands.
Crystal structures
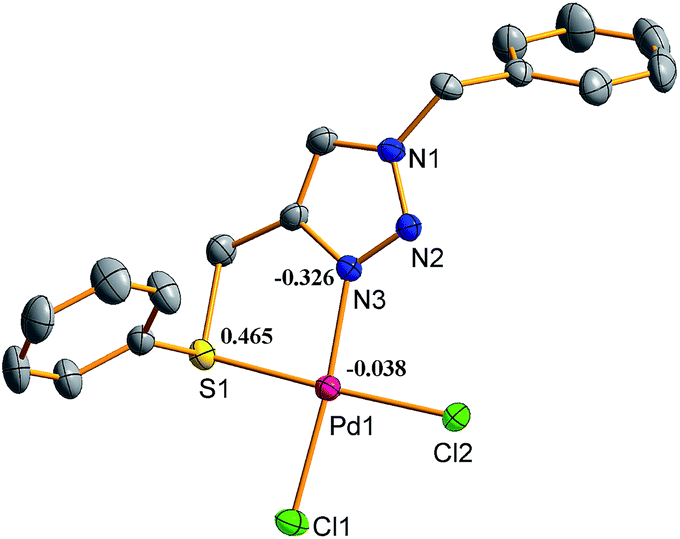
The crystal structures of 1 and 2 have been solved. The crystallographic data and refinement parameters are given in ESI (Table S1†). The ORTEP diagrams of 1 and 2 with 30% probability ellipsoids are shown in Fig. 1 and 2 with selected bond distances and angles.
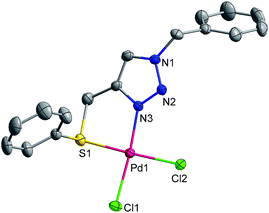 |
| | Fig. 1 Molecular structure of complex 1. Bond length (Å): N(3)–Pd(1) 2.005(4), Pd(1)–S(1) 2.2727(14), Cl(1)–Pd(1) 2.2968(14), Cl(2)–Pd(1) 2.3003(13). Bond angle [°]: N(3)–Pd(1)–Cl(1) 174.04(11), N(3)–Pd(1)–Cl(2) 92.72(11), S(1)–Pd(1)–Cl(1) 89.01(5), S(1)–Pd(1)–Cl(2) 177.91(5), Cl(1)–Pd(1)–Cl(2) 93.09(5), N(3)–Pd(1)–S(1) 85.18(11). | |
 |
| | Fig. 2 Molecular structure of complex 2. Bond length (Å): Pd(1)–N(3) 2.005(4), Pd(1)–Cl(1) 2.2799(15), Pd(1)–Cl(2) 2.3146(16), Pd(1)–Se(1) 2.3693(8). Bond angle [°]: N(3)–Pd(1)–Cl(1) 173.41(14), N(3)–Pd(1)–Cl(2) 95.32(14), N(3)–Pd(1)–Se(1) 85.37(13), Cl(1)–Pd(1)–Se(1) 88.11(4), Cl(2)–Pd(1)–Se(1) 178.20(5), Cl(1)–Pd(1)–Cl(2) 91.22(6). | |
H atoms are omitted for clarity. More bond angles and distances are given in ESI (Table S2†). The L1 and L2 exhibit identical bonding mode in both the complexes 1 and 2, that is, a five-membered chelate ring is formed by their coordination to the metal centre. In both complexes 1 and 2 palladium has distorted square-planar geometry. The S and Se are at a cis position to N and two chlorides are also cis to each other. The Pd–S bond length in 1 [2.2727(14) Å] is somewhat larger than that of palladacycle formed with 2,3-bis[(phenylthio)methyl]quinoxaline by activation of C(sp3)–H [2.259(2) Å],43 and consistent with the value reported for Pd(II) complex of sulphated Schiff base of 1-hydroxy-2-acetophenone [2.2704(16) Å].41 In complex 2, the Pd–Se bond length [2.3693(8) Å] is close to the value reported for Pd(II) complex of a selenated Schiff base of 1-hydroxy-2-acetophenone [2.3600(15) Å],41 and somewhat shorter than that of Pd(II) complex of (Se, N, Se) pincer ligand [2.3891(7) Å].48 The Pd–N bond length in both the complexes 1 and 2 are same [2.005(4) Å] and comparable to the value reported for Pd(II) complex of a click generated (P, C, S) pincer ligand [2.015(19) Å].50 The Pd–Cl bond lengths of 1 [2.2968(14)–2.3003(13) Å] and 2 [2.2799(15)–2.3146(16) Å] are in the normal range.43
The non-covalent secondary C–H⋯Cl interactions in the crystals of complexes 1 and 2 are shown in Fig. 3 and 4. They result in the formation of a sheet like structure. These C–H⋯Cl distances are given in the ESI (Table S3†).
 |
| | Fig. 3 Non covalent C–H⋯Cl interaction in 1. | |
 |
| | Fig. 4 Noncovalent C–H⋯Cl interaction in 2. | |
Catalytic studies
Complexes 1 and 2 have been explored as catalysts for Suzuki–Miyaura and Heck C–C coupling reactions of several aryl bromides including electronically deactivated ones. The results of catalysis of SMC and HC with complexes 1 and 2 are given in Table 1 and 2 respectively. These results suggest that the two complexes are more efficient for Suzuki coupling than Heck, as yields of cross-coupled products are high in SMC at a relatively lower loading of catalyst. The deactivated substrates give good yields in Suzuki coupling (Table 1: entries 7 and 8). However, Heck coupling of 4-bromoanisole catalyzed with 1 or 2 does not result in coupled product. The efficiency of catalyst 1 has been found marginally higher (Tables 1 and 2) than that of its selenium analogue 2. However, it can not be generalized, as sulfur ligands provide more reactive complexes than their selenium analogues. The difference in yields of coupled products in the reactions catalyzed with 1 and 2 probably does not arise due to work up.
Table 1 Suzuki–Miyaura coupling reaction catalyzed by complex 1 and 2a

|
| Entry |
Aryl halide |
Catalyst 1 |
Catalyst 2 |
| Mol% |
t (h) |
Yieldb/c |
Mol% |
t (h) |
Yieldc |
| Reaction conditions: 1.0 equiv. of ArBr, 1.5 equiv. of phenylboronic acid, and 2 equiv. of K2CO3, solvent: water + DMF, and bath temperature 90 °C. Isolated yield after column chromatography. NMR yield%. |
| 1 |
4-Bromobenzaldehyde |
0.01 |
3 |
95/100 |
0.01 |
6 |
92 |
| 2 |
4-Bromobenzonitrile |
0.01 |
3 |
85/91 |
0.01 |
6 |
85 |
| 3 |
4-Bromonitrobenzene |
0.01 |
12 |
92/97 |
0.01 |
12 |
87 |
| 4 |
4-Bromoacetophenone |
0.01 |
3 |
90/95 |
0.05 |
3 |
78 |
| 5 |
4-Bromobenzoic acid |
0.1 |
6 |
82/87 |
0.1 |
6 |
80 |
| 6 |
Bromobenzene |
0.1 |
3 |
75/80 |
0.1 |
12 |
88 |
| 7 |
4-Bromotoluene |
0.1 |
3 |
85/91 |
0.1 |
3 |
82 |
| 8 |
4-Bromoanisole |
0.1 |
3 |
90/96 |
0.1 |
6 |
88 |
Table 2 Heck coupling reaction catalyzed by complex 1 and 2a
|
| Entry |
Aryl halide |
Catalyst 1 |
Catalyst 2 |
| Mol% |
t (h) |
Yieldb/c |
Mol% |
t (h) |
Yieldc |
| Reaction conditions: 1.0 equiv. of ArBr, 1.0 equiv. of n-butyl acrylate, and 2 equiv. of base (K2CO3), solvent DMF and bath temperature 110 °C, N2 atm. Isolated yield after column chromatography. NMR yield%. |
| 1 |
4-Bromobenzaldehyde |
0.01 |
12 |
95/100 |
0.01 |
12 |
92 |
| 2 |
4-Bromobenzonitrile |
0.1 |
12 |
90/94 |
0.1 |
12 |
88 |
| 3 |
4-Bromonitrobenzene |
0.1 |
12 |
92/97 |
0.1 |
12 |
88 |
| 4 |
4-Bromoacetophenone |
0.1 |
24 |
78/83 |
0.1 |
36 |
65 |
| 5 |
4-Bromobenzoic acid |
1 |
24 |
85/90 |
1 |
24 |
82 |
| 6 |
Bromobenzene |
0.5 |
24 |
75/81 |
0.5 |
36 |
78 |
| 7 |
4-Bromotoluene |
1 |
36 |
70/75 |
1 |
36 |
68 |
| 8 |
4-Bromoanisole |
1 |
24 |
—/— |
1 |
24 |
— |
In the course of both the coupling reactions, black particles appear. This indicates that complexes 1 and 2 are probably pre-catalysts and dispense black particles i.e. Pd-containing species in the course of reaction which appear to have some role in the catalytic process.
The particles, formed from 1 and 2 during catalysis, under optimum conditions, of Suzuki coupling between 4-bromo-benzonitrile and phenylboronic acid and Heck coupling between 4-bromobenzaldehyde and n-butyl acrylate were isolated. The SEM, SEM-EDX and HR-TEM of these particles have indicted them as nanosized spheres. Their sizes vary with the catalyst and coupling reaction. In Suzuki coupling catalyzed with 1, NPs of size 3–5 nm were obtained, whereas in case of 2 they were of 5–7 nm (Fig. 5). The sizes of NPs obtained in Heck reaction activated with complexes 1 and 2 were found in ranges 5–6 and 9–11 nm (Fig. 6) respectively. Further SEM-EDX has revealed that the NPs generated from both the coupling reaction are composed of palladium and sulphur or selenium. The Pd:S ratio in NPs generated in SMC catalyzed with 1 is 64:36 whereas in the case of 2, the Pd:Se ratio in NPs has been found to be 61:39 (ESI: Fig. S12 and S13†). However, Pd:S or Se ratio in NPs generated in Heck coupling activated with 1 and 2 are respectively 64:36 and 85:15 (ESI: Fig. S14 and S15†). All NPs were subjected to TGA upto 700 °C (ESI: Fig. S16–S19†). The weight losses shown by NPs obtained from complexes 1 and 2 in SMC, are 35 and 28% respectively. The weight losses from NPs formed in catalytic Heck coupling with the two complexes have been found to be 21 and 48% respectively. These weight losses appear due to the presence of organic contents with nanoparticles.
 |
| | Fig. 5 HRTEM image of NPs obtained from catalyst 1(a) and 2(b) respectively during SMC (scale bar 20 and 50 nm). | |
 |
| | Fig. 6 HRTEM image of NPs obtained from complex 1(c) and 2(d) respectively during Heck coupling (scale bar 20 and 50 nm). | |
To understand further the actual phase of NPs formed in the catalytic processes, the black residues obtained from complexes 1 and 2 in Suzuki and Heck coupling reactions, were annealed at temperatures at which their weight losses were found maximum in TGA (ESI: Fig. S16–S19†). These annealed residues were subjected to PXRD, SEM and TEM analysis. In PXRD there are three intense lines in each case with a feeble peak in case of NPs given by 2. The d-spacing corresponding to three intense PXRD lines in all the cases are 2.24, 1.94 and 1.37 Å, which correspond to (111), (200) and (220) planes with lattice constant, a = 3.882 Å, matching with that of JCPDS file 01-1201 of Pd metal. The low intensity peak at 2.56 Å in PXRD of residues obtained from complex 2 in Suzuki and Heck coupling corresponds to (120) plane of selenium (not a part of any selenide phase) and indicates the presence of low crystalline discrete selenium (ESI: Fig. S20–S23†). The SEM-EDX analyses of annealed NPs obtained from complex 1 (ESI: Fig. S28–S31†) indicate the presence of palladium and sulfur in them in ratios: 45:55 and 82:18 for Suzuki and Heck reaction respectively. Palladium and selenium ratios for annealed NPs of complex 2 are 73:27 and 41:59 for Suzuki and Heck coupling respectively. Most probably S is also present as discrete species like Se but being non-crystalline does not appear in PXRD. TEM images suggest that the NPs obtained from complex 1 in Suzuki coupling, are grown to the size of ∼120–200 nm on annealing whereas size of those obtained from 2 grows upto ∼60–90 nm only (Fig. 7). The sizes of NPs obtained in Heck reactions activated with complexes 1 and 2 after annealing have been found in the ranges 20–60 and 60–75 nm respectively (Fig. 8).
 |
| | Fig. 7 TEM image of NPs obtained from complex 1(e) and 2(f) respectively after annealing after Suzuki coupling (scale bar 1 μm and 100 nm). | |
 |
| | Fig. 8 TEM image of NPs obtained from complex 1 and 2 respectively after annealing after Heck coupling (scale bar 50 and 100 nm). | |
These isolated palladium-NPs (before annealing) protected with a chalcogen ligand or its fragments were found to catalyze Suzuki–Miyaura and Heck coupling independently, indicating their role in the catalytic process. 4-Bromobenzaldehyde and 4-bromoacetophenone couple smoothly (Table 3) with n-butyl acrylate or PhB(OH)2 in DMF or DMF + H2O mixture (3.0 + 2.0 mL) in the presence of the NPs obtained from 1 or 2. However, yields were relatively less when their coupling reactions were activated with NPs obtained from 2. The activities of nanoparticles obtained from 1 and 2 were found very different in the case of SMC of 4-bromoanisole, which gave cross-coupled product in 75% yield in the presence of NPs obtained from 1 but did not show any conversion with NPs of 2. On isolation of NPs diminishing effect on their catalytic efficiency relative to in situ generated ones, can be noticed but is understandable due to possible aggregation.46
Table 3 Suzuki Miyauraa and Heck couplingb reactions catalyzed by isolated NPs obtained from complexes 1 and 2
| Entry |
Aryl halide |
Yieldc (%) |
| Suzuki coupling |
Heck coupling |
| NPs obtained from complex 1 during SMCd |
NPs obtained from complex 2 during SMCe |
NPs obtained from complex 1 during Heckf |
NPs obtained from complex 2 during Heck couplingg |
| Reaction conditions: 1.0 equiv. of ArBr, 1.5 equiv. of phenylboronic acid, and 2 equiv. of base (K2CO3), DMF + water as solvent and bath temperature 90 °C. Time: 3 (SMC) and 10 h (HC) 1.0 equiv. of ArBr, 1 equiv. of n-butyl acrylate, and 2 equiv. of base (K2CO3), DMF as solvent, bath temperature 110 °C, N2 atm. NMR yield%. 5 mg of NPs. 30 mg of NPs. 30 mg of NPs. 50 mg of NPs. |
| 1 |
4-Bromo-benzaldehyde |
94 |
85 |
75 |
70 |
| 2 |
4-Bromo-acetophenone |
90 |
60 |
70 |
65 |
| 3 |
4-Bromo-anisole |
75 |
— |
— |
— |
The independently tested activities of NPs strongly support their role in catalysis but does not rule out the involvement of molecular Pd(II) complex in catalysis. Thus mercury and PPh3 poisoning experiments were carried out.51 The presence of excess of elemental Hg has been found to suppress completely the catalysis in both reactions; Suzuki coupling of phenylboronic acid with 4-bromoacetophenone (0.5 mol% loading of 1) and 4-bromobenzaldehyde (0.1 mol% loading of 2) and Heck reaction between 4-bromobenzaldehyde and n-butyl acrylate at 1 mol% loading of 1 or 2. The PPh3 poisoning test was also positive like Hg poisoning test. These results indicate that catalysis is carried out primarily by in situ generated palladium NPs and direct role of discrete molecular Pd(II) complex in catalysis is less, as molecular complex functioning in a homogeneous manner containing metal in a high oxidation state that is tightly bound by protective ligands, cannot be poisoned.
The in situ generated NPs of palladium chalcogenides or palladium protected with organochalcogen moieties may contribute in a homogeneous52,53 as well as heterogeneous fashion.54 Both the modes of catalysis have been reported in case of preformed NPs. To study the possibility of homogeneous catalysis by NPs in the present case, a two phase test was carried out (Scheme 2) for both Suzuki and Heck coupling reactions activated by complex 1. This is a modified version of “three-phase test” proposed originally by Rebek and co-workers55,56 and used by Davies et al.57 and others38 for solid phase catalysts.
 |
| | Scheme 2 Two phase test in Suzuki and Heck coupling. | |
The covalently immobilized aryl bromide onto silica surface and free ArBr were taken as substrates and subjected to Suzuki and Heck coupling (Scheme 2). The soluble part was separated by filtration and analyzed after workup with 1H NMR to determine the yield of the cross-coupled product, which was found to be 95% (Scheme 2) in case of Suzuki coupling and 75% in case of Heck coupling (Scheme 2). Thereafter, the solid phase was hydrolyzed and after workup analysed for the resulting product with 1H NMR. The 70–72% of the immobilized 4-bromobenzoic acid (as amide) was converted to the cross-coupled product in case of both the coupling reactions. This indicates that there is a major contribution by molecular or colloidal Pd species generated from NPs, as a homogeneous catalyst. These species include palladium atoms leached from the in situ generated Pd containing NPs and/or the molecular species.58 If activity of the catalyst is fully heterogeneous in nature, the anchored aryl bromide is not expected to be converted into a cross-coupled product. The heterogeneous process also exists as 28–30% immobilized substrate remains unchanged. Simultaneous presence of homogeneous and heterogeneous process has been recently described as a “cocktail”-like catalysis.58 The present complexes 1 and 2 as a catalyst for Suzuki coupling perform better than some Pd complexes of ligands having similar donor sites. For example less loading and reaction time is required in the present case in comparison to palladium(II) complexes of sulphated of selenated Schiff bases of salicylaldehyde.5b On comparing carbothioamide derived palladacycles5c with 1, the performance of the latter appears to be better in both SMC and HC. However, complex [PhN = C(CMe2)(N-η1-Ph)C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (CH2)2SR]Pd(OAc) [R = CMe3; R = Ph]4b gives better conversions for both SMC and HC in comparison to those of 1. The performance of 2 for HC is comparable to a Pd-complex of (S, N) ligand.4c
N (CH2)2SR]Pd(OAc) [R = CMe3; R = Ph]4b gives better conversions for both SMC and HC in comparison to those of 1. The performance of 2 for HC is comparable to a Pd-complex of (S, N) ligand.4c
DFT calculations
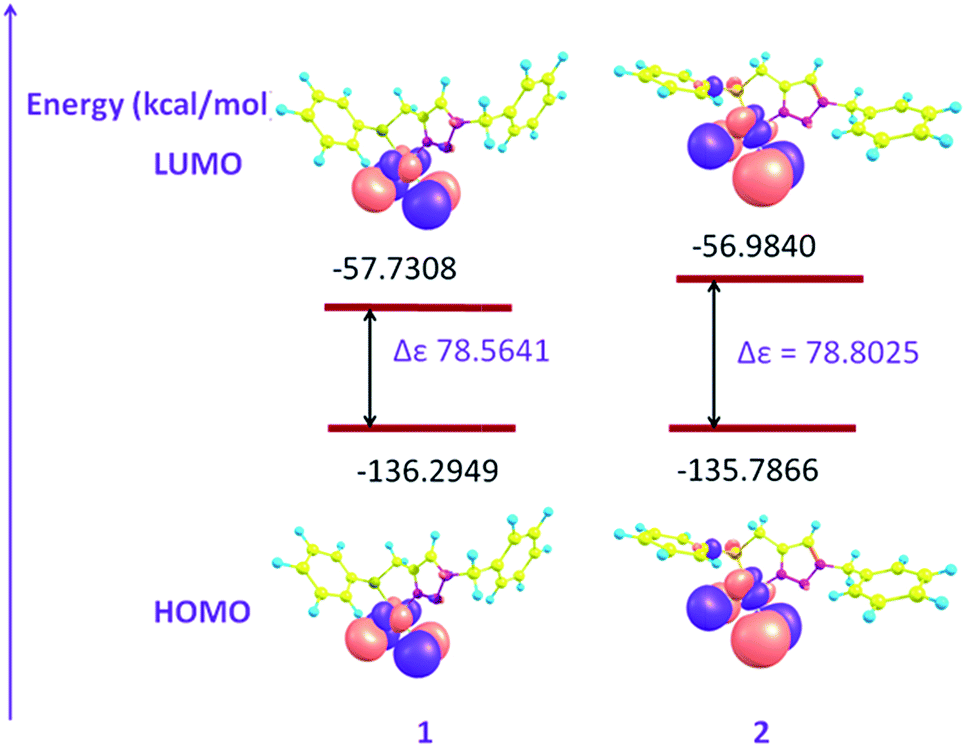
The density functional theory (DFT) calculations were performed on both the complexes to rationalize the nature of bonding within these complexes and their reactivity in terms of energy of molecular orbitals. In both the complexes the HOMOs (highest occupied molecular orbitals), constituted by interaction of a d orbital of Pd(II) and a p orbital of chlorine atom, are positioned primarily over the Pd and Cl atoms. In HOMO-1 of both the complexes metal d orbital, p-orbitals of a chalcogen donor atom, chlorine atoms and one of the nitrogen of triazole ring have been found to overlap with each other. The chemical reactivity of a complex and its HOMO–LUMO energy gap may be correlated. The high reactivity of a complex may be at least partially ascribed to low value of this energy gap and increment in the HOMO–LUMO energy gap results in the reduction of reactivity. Therefore among the two complexes, complex 1 which has lower HOMO–LUMO energy gap (Fig. 9) is supposed to be better catalyst than 2, as found experimentally. The electron densities at Se, Pd and N(3) of triazole ring are high in case of 2 (Fig. 10 and 11) which probably result in strong coordination of donor atoms with metal in 2 and in turn its higher stability relative to 1.
 |
| | Fig. 9 Frontier molecular orbital diagrams of complexes 1 and 2. | |
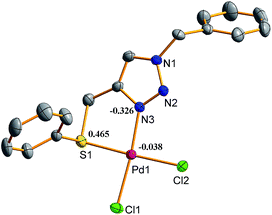 |
| | Fig. 10 Mulliken partial charges of complex 1. | |
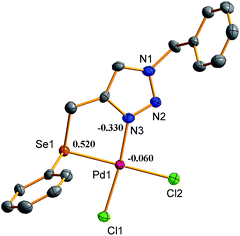 |
| | Fig. 11 Mulliken partial charges of complex 2. | |
Therefore, probably the complex 2 being more stable is less reactive than 1.29,59,60 There is a good match between experimentally observed and calculated distances for Pd–Cl and Pd–N bonds (ESI: Table S4†). However, some variations between calculated and observed Pd–E (E = S/Se) bond distances (0.196–0.240 Å) exist. The calculated and experimentally found bond angles are also very close (ESI: Table S4†) to each other.
Conclusions
Palladium(II) complexes [Pd(L)Cl2] (1 and 2) of 1-benzyl-4-((phenylthio)-/(phenylseleno)methyl)-1H-1,2,3-triazole (L1/L2 = L) have been synthesized and found suitable as catalyst at their low loading (0.01 mol%) for SMC and HC of various aryl bromides including deactivated ones. The catalytic processes are more efficient with 1 in which palladium is coordinated with sulfur. The catalysis of both the coupling reactions occurs via in situ generated nanoparticles of size 3–11 nm composed of palladium and sulfur or selenium and protected by L1 or L2 or its fragments. In case of Heck coupling most probably the role of Pd based NPs has been established so explicitly for the first time. The results of a two-phase test suggest that the catalysis is cocktail type. The catalytic activity of these complexes has also been explained in the term HOMO–LUMO energy gaps. Complex 1 having low HOMO–LUMO energy gap and low electron density on S, N(3) and Pd shows higher activity than that of 2.
Experimental section
Chemicals and reagents
The ligands L1 and L2 were synthesized as reported earlier.29 The PdCl2 and aryl bromides were procured from Sigma-Aldrich (USA) and used without any further purification. The [(CH3CN)2PdCl2] was prepared according to reported method.61 Common reagents available locally were used. Solvents for synthesis were dried and distilled before use by conventional procedures.42
Physical measurement
The 1H, 13C{1H}, and 77Se{1H} NMR spectra were recorded on a Bruker Spectrospin DPX-300 NMR spectrometer at 300.13, 75.47, and 57.24 MHz, respectively with chemical shifts reported in ppm relative to normal standards. Yields refer to isolated yields of compounds which have purity ≥95% [established by 1H NMR]. All reactions were carried out in glassware dried in an oven, under ambient conditions. The commercial nitrogen gas was used after passing it successively through traps containing solutions of alkaline anthraquinone, sodium-dithionite, alkaline pyrogallol, concentrated H2SO4 and KOH pellets. Nitrogen atmosphere if required was created using Schlenk techniques. IR spectra in the range 4000–400 cm−1 were recorded on a Nicolet Protége 460 FT-IR spectrometer as KBr pellets. The C, H and N analyses were carried out with a Perkin-Elmer 2400 Series II C, H and N analyser. Single-crystal structure data were collected with a Bruker AXS SMART Apex CCD diffractometer using Mo Kα (0.71073 Å) radiation at 298(2) K. The software SADABS was used for absorption correction (if needed) and SHELXTL for space group, structure determination, and refinements. Hydrogen atoms were included in idealized positions with isotropic thermal parameters set at 1.2 times that of the carbon atom to which they were attached in all cases. The least-square refinement cycles on F2 were performed until the model converged. The melting points determined in an open capillary are reported as such. High resolution mass spectral (HR-MS) measurements were performed with electron spray ionization (10 eV, 180 °C source temperature and sodium formate as calibrant) on a Bruker MIcroTOF-Q II, taking sample in CH3CN. High-resolution TEM characterizations were carried out with a Technai G2 20 electron microscope operated at 200 kV. The specimens for HR-TEM were prepared by dispersing the powdered sample in chloroform by ultrasonic treatment, dropping the slurry onto a porous carbon film supported on a copper grid, and then drying it in air. The phase morphologies of the samples were studied using a Carl Zeiss EVO5O Scanning Electron Microscope (SEM). Samples were mounted on a circular metallic sample holder with a sticky carbon tape. Elemental compositions of nano-particles on SEM were analyzed by EDX system based on the SDD technology, (model: Quan Tax 200 with an energy resolution of 127 eV at Mn Kα or Quanta 200 FEG with Oxford-EDS system IE250X-MAX80). The samples were scanned in different regions in order to minimize the error in the analysis for evaluating the morphological parameters. Thermogravimetric analysis (TGA) was carried out using a Perkin-Elmer system in a flowing nitrogen atmosphere with a heating rate of 10 °C min−1. All DFT calculations have been carried out at the Department of Chemistry, Supercomputing Facility for Bioinformatics and Computational Biology, IIT Delhi, with the GAUSSIAN-03 (ref. 62) programs. The geometry of complexes 1 to 2 has been optimized at the B3LYP63 level using a SDD basis set for metal atoms and S and Se and 6-31G* basis sets for C, N and H. Geometry optimizations have been carried out without any symmetry restriction with X-ray coordinates of the molecule. Harmonic force constants have been computed at the optimized geometries to characterize the stationary points as minima. The molecular orbital plots have been made using the Chemcraft program package (http://www.chemcraftprog.com).
Synthesis of complexes 1 and 2
To a solution of L1 (0.056 g, 0.2 mmol) or L2 (0.065 g, 0.2 mmol) in THF (2 mL) was added a solution of [(CH3CN)2PdCl2] (0.052 g, 0.2 mmol) in THF (2 mL). The reaction mixture was stirred for 2 h at room temperature. The development of pale yellow colour indicated the formation of complex. The solvent was removed under reduced pressure and the residue was washed with ether (5 mL) and dried in vacuo. Single crystal of 1 and 2 were obtained from a mixture (1:1) of CH3OH and CH3CN.
1. Yield: 0.082 g (90%). M.p. 200 °C (d). Anal. found: C, 41.86; H, 3.26; N, 9.20%. Calcd for C16H15N3Cl2PdS: C, 41.89; H, 3.30; N, 9.16%.1H NMR (DMSO-d6, 25 °C, TMS) δ (ppm): 4.20–4.65 (m, 2H, H5), 5.64 (s, 2H, H8), 7.14–7.44 (m, 9H, H1–H3, H10, H11), 7.72 (s, 1H, H12), 8.36 (s, 1H, H7). 13C{1H} NMR (DMSO-d6, 25 °C, TMS) δ (ppm): 35.1 (C5), 54.9 (C8), 124.4 (C12), 127.7 (C11), 128.7 (C3), 128.8 (C2), 128.9 (C10), 129.5 (C9), 130.1 (C1), 130.6 (C7), 134.5 (C4), 150.4 (C6). IR (cm−1): 438 (b), 680 (m), 748 [s; C–H (aromatic) bending], 1035 (m), 1144 (m), 1253 (m), 1349 (m), 1444 [s; νC–C (aromatic)], 1568 [b; νCN], 1643 [m; νNN], 1703 (s), 2358 (m), 2917 [m; νC–H (aliphatic)], 3083 [m; νC–H (aromatic)], 3132 (m). HR-MS (CH3CN) [M + Na]+ (m/z) = 479.9304; calcd value for C16H15N3Cl2NaPdS = 479.9291 (δ 2.7 ppm).
2. Yield: 0.085 g (85%). M.p. 220 °C (d). Anal. found: C, 38.06; H, 2.96; N, 8.35%. Calcd for C16H15N3Cl2PdSe: C, 38.01; H, 2.99; N, 8.31%. 1H NMR (DMSO-d6, 25 °C, TMS) δ (ppm): 4.18 (d, 1H, 3JH–H = 19.2 Hz, H5), 4.56 (d, 1H, 3JH–H = 12 Hz, H5), 5.68 (s, 2H, H8), 7.28–7.48 (m, 8H, H1, H2, H10–H12), 8.07 (m, 2H, H3), 8.37 (s, 1H, H7). 13C{1H} NMR (DMSO-d6, 25 °C, TMS) δ (ppm): 28.3 (C5), 55.2 (C8), 125.4 (C12), 128.2 (C4), 128.5 (C3), 129.1 (C7), 129.4 (C1), 129.4 (C2), 130.5 (C11), 132.3 (C10), 134.9 (C9), 152.1 (C6). 77Se{1H} NMR (DMSO-d6, 25 °C, Me2Se) δ (ppm): 460. IR (cm−1): 464 (m), 688 (s), 741 [s; C–H (aromatic) bending], 1030 (m), 1241 (m), 1354 (m), 1398 (s), 1445 [b; νC–C (aromatic)], 1564 [b; νCN], 1646 [b; νNN], 2362 (s), 2928 [m; νC–H (aliphatic)], 3080 [m; νC–H (aromatic)], 3117 (m). HR-MS (CH3CN) [M + Na]+ (m/z) = 527.8730; calcd value for C16H15N3Cl2PdNaSe = 527.8738 (δ 0.9 ppm).
Procedure for the Suzuki reaction of aryl halides with phenylboronic acid
An oven-dried flask was charged with aryl bromide (1.0 mmol), phenylboronic acid (1.5 mmol), K2CO3 (2.0 mmol), DMF–H2O (3.0 mL/2.0 mL) and catalyst 1 or 2 (0.01–0.1 mol%). The flask was placed on an oil bath at 90 °C under aerobic conditions and the reaction mixture was stirred until maximum conversion of aryl halide to product occurred. The mixture was extracted with diethylether, washed with water and dried over anhydrous Na2SO4. The solvent of the extract was evaporated off on a rotary evaporator, and the residue was purified by a column of silica gel using CHCl3–hexane as eluent. The products were authenticated by 1H and 13C{1H} NMR spectra.
Procedure for Heck reaction of aryl halides with n-butyl acrylate
A mixture of n-butyl acrylate (1 mmol), aryl halide (1 mmol), DMF (3 mL), and complex 1 or 2 (0.01–1 mol%) was placed on oil bath maintained at 110 °C under inert atmosphere and the reaction mixture was stirred until maximum conversion of aryl halide to product occurred. It was cooled to room temperature and the product was extracted with ethyl acetate. The extract was washed with water and dried over anhydrous Na2SO4. Its solvent was evaporated on a rotary evaporator to obtain the product, which was purified by a column of silica gel using CHCl3–hexane as eluent. The products were authenticated by 1H and 13C{1H} NMR spectra.
Isolation of nanoparticles generated from 1 and 2 during Suzuki coupling
A mixture of Pd(II) complex 1 or 2 (0.5 mmol), phenylboronic acid (1.5 mmol), 4-bromobenzonitrile (1.0 mmol) and K2CO3 (2.0 mmol) in DMF (3.0 mL) and water (2.0 mL) was heated at 90 °C for 3 h and then cooled to room temperature. The solvent was decanted and the black residue (NPs) was thoroughly washed with water–acetone mixture (1:3) and dried in vacuo.
Isolation of nanoparticles generated from 1 and 2 during Heck coupling
A mixture of Pd(II) complex 1 or 2 (0.5 mmol), n-butyl acrylate (1 mmol), 4-bromobenzaldehyde (1 mmol), DMF (3 mL), was placed on an oil bath at 110 °C in an inert atmosphere for 3 h and then cooled to room temperature. The solvent was decanted and the black residue (NPs) was thoroughly washed with water–acetone mixture (1:3) and dried in vacuo.
Procedure for the Suzuki–Miyaura and Heck coupling reaction catalysed by NPs generated from 1 and 2
It was similar to the one used for complexes 1 and 2 except the reaction time which was 3 h for Suzuki reaction and 10 h for Heck. The amount of NPs used for Suzuki reaction was 5 (for 1) and 30 mg (for 2), whereas for Heck coupling the amounts were 30 and 50 mg respectively.
Hg poisoning test
An excess of Hg (Hg:Pd: 500:1) was taken in the reaction flask and the coupling reaction carried out. Suzuki coupling of 4-bromoacetophenone was tested under optimum reaction conditions at 0.5 mol% loading of complex 1, whereas 4-bromobenzaldehyde was chosen as substrate in case of 2 with a loading of 0.1 mol%. Heck coupling between 4-bromobenzaldehyde and n-butyl acrylate was tested at 1 mol% loading of 1 or 2. The reaction mixtures were analyzed after 3 h in all the cases to study the yields of cross-coupled products and it was found that no reaction took place.
PPh3 test
It was carried in a manner similar to that of Hg poisoning test using identical substrates except the amount of PPh3 which was 100 equivalents with respect to catalyst. No cross-coupled product was obtained even after 3 h of reaction.
Two phase test
A mixture of 4-bromobenzoic acid immobilized on silica as amide (0.20 g) prepared by standard procedure,64 phenylboronic acid (0.36 g, 3 mmol) or n-butyl acrylate (2 mmol), 4-bromoacetophenone (0.20 g, 1 mmol), K2CO3 (0.56 g, 4 mmol) and catalyst (see Scheme 2) were taken in a mixture of DMF and water (8 + 4 mL) or 8 mL of DMF (Scheme 2). The mixture was heated for 12 h at 90 °C in case of Suzuki coupling and 110 °C in case of Heck. It was cooled and filtered through G-4 crucible. The residue was washed with 20 mL of H2O followed by diethylether (50 mL). The filtrate and washings were collected together and mixed with 50 mL of water. The resulting mixture was extracted with diethylether (50 mL). The solvent of the extract was evaporated off on a rotary evaporator and residue subjected to 1H NMR. The solid residue was hydrolyzed with KOH (1.68 g dissolved in 2:1 EtOH–H2O mixture at 90 °C) for 3 days. The resulting solution was neutralized with aqueous 20% (v/v) HCl, extracted with dichloromethane (2 × 20 mL) followed by ethyl acetate (2 × 15 mL). The solvent of combined extract was evaporated off and the resulting residue was analyzed with 1H NMR.
Acknowledgements
Council of Scientific and Industrial Research (CSIR), India supported the work through the award of SRF/RA to GKR and AK and project 01(2421)10/EMR-II. FS, SK and MPS thank to UGC for fellowship. Authors thank Professor A. K. Ganguli of IIT Delhi for providing HR-TEM facility. Authors also thank Prof. B. Jayaram (Coordinator, SCFBio) and Goutam Mukherjee for the Supercomputing Facility for Bioinformatics and Computational Biology (SCFBio), Department of Chemistry, IIT Delhi, for providing access to the computational facilities.
Notes and references
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed.
- I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS PubMed.
- A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS PubMed.
-
(a) D. Mc Cartney and P. J. Guiry, Chem. Soc. Rev., 2011, 40, 5122 RSC;
(b) M.-T. Chen, C.-A. Huang and C.-T. Chen, Eur. J. Inorg. Chem., 2006, 4642 CrossRef CAS;
(c) T. Chakraborty, K. Srivastava, H. B. Singh and R. J. Butcher, J. Organomet. Chem., 2011, 696, 2559 CrossRef CAS PubMed.
-
(a) S. R. Chemler, D. Trauner and S. J. Danishefsky, Angew. Chem., Int. Ed., 2001, 40, 4544 CrossRef CAS;
(b) I. D. Kostas, B. R. Steele, A. Terzis, S. V. Amosova, A. V. Martynov and N. A. Makhaeva, Eur. J. Inorg. Chem., 2006, 2642 CrossRef CAS;
(c) Z. Xiong, N. Wang, M. Dai, A. Li, J. Chen and Z. Yang, Org. Lett., 2004, 6, 3337 CrossRef CAS PubMed.
- R. Rossi, F. Bellina and M. Lessi, Adv. Synth. Catal., 2012, 354, 1181 CrossRef CAS.
- F. Bellina, A. Carpita and R. Rossi, Synthesis, 2004, 15, 2419 Search PubMed.
- H. Li, C. C. C. J. Seechurn and T. J. Colacot, ACS Catal., 2012, 2, 1147 CrossRef CAS.
- N. T. S. Phan, M. V. D. Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609 CrossRef CAS.
- R. Narayanan, Molecules, 2010, 15, 2124 CrossRef CAS PubMed.
- A. Fihri, M. Bouhrara, B. Nekoueishahraki, J.-M. Basset and V. Polshettiwar, Chem. Soc. Rev., 2011, 40, 5181 RSC.
- D. Astruc, Inorg. Chem., 2007, 46, 1884 CrossRef CAS PubMed.
- M. T. Reetz and J. G. de Vries, Chem. Commun., 2004, 1559 RSC.
- J. G. de Vries, Dalton Trans., 2006, 421 RSC.
- A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176 CrossRef CAS.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CrossRef CAS PubMed.
- H. Turkmen, R. Can and B. Cetinkaya, Dalton Trans., 2009, 7039 RSC.
- C. A. Fleckenstein and H. Plenio, Chem. Soc. Rev., 2010, 39, 694 RSC.
- J. P. Wolfe and S. L. Buchwald, Angew. Chem., Int. Ed., 1999, 38, 2413 CrossRef CAS.
- A. F. Littke, C. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020 CrossRef CAS.
- X. Bei, H. W. Turner, W. H. Weinberg and A. S. Guram, J. Org. Chem., 1999, 64, 6797 CrossRef CAS PubMed.
- R. B. Bedford and C. S. J. Cazin, Chem. Commun., 2001, 1540 RSC.
- R. B. Bedford, S. L. Hazelwood and M. E. Limmert, Chem. Commun., 2002, 2610 RSC.
- C. A. Fleckenstein and H. Plenio, Organometallics, 2007, 26, 2758 CrossRef CAS.
-
(a) X. Cui, J. Li, Z.-P. Zhang, Y. Fu, L. Liu and Q.-X. Guo, J. Org. Chem., 2007, 72, 9342 CrossRef CAS PubMed;
(b) X. Cui, Z. Li, C.-Z. Tao, Y. Xu, J. Li, L. Liu and Q.-X. Guo, Org. Lett., 2006, 12, 2467 CrossRef PubMed;
(c) A. Fihri, P. Meunier and J.-C. Hierso, Coord. Chem. Rev., 2007, 251, 2017 CrossRef CAS PubMed.
- D. E. Bergbreiter, P. L. Osburn and Y.-S. Liu, J. Am. Chem. Soc., 1999, 121, 9531 CrossRef CAS.
- D. E. Bergbreiter, P. L. Osburn, A. Wilson and E. M. Sink, J. Am. Chem. Soc., 2000, 122, 9058 CrossRef CAS.
- D. E. Bergbreiter, P. L. Osburn and J. D. Frels, J. Am. Chem. Soc., 2001, 123, 11105 CrossRef CAS.
- F. Saleem, G. K. Rao, A. Kumar, G. Mukherjee and A. K. Singh, Organometallics, 2013, 32, 3595 CrossRef CAS.
- F. Saleem, G. K. Rao, A. Kumar, G. Mukherjee and A. K. Singh, Organometallics, 2014, 33, 2341 CrossRef CAS.
- A. Kumar, G. K. Rao, F. Saleem and A. K. Singh, Dalton Trans., 2012, 41, 11949 RSC.
- A. Kumar, G. K. Rao and A. K. Singh, RSC Adv., 2012, 2, 12552–12574 RSC.
- G. K. Rao, A. Kumar, M. P. Singh and A. K. Singh, J. Organomet. Chem., 2014, 753, 42 CrossRef CAS PubMed.
- A. Kumar, G. K. Rao, S. Kumar and A. K. Singh, Dalton Trans., 2013, 42, 5200 RSC.
- G. K. Rao, A. Kumar, M. P. Singh and A. K. Singh, J. Organomet. Chem., 2014, 749, 1 CrossRef CAS PubMed.
- A. Kumar, G. K. Rao, F. Saleem, R. Kumar and A. K. Singh, J. Hazard. Mater., 2014, 269, 9 CrossRef CAS PubMed.
- G. K. Rao, A. Kumar, M. Bhunia, M. P. Singh and A. K. Singh, J. Hazard. Mater., 2014, 269, 18 CrossRef CAS PubMed.
- A. Kumar, G. K. Rao, S. Kumar and A. K. Singh, Organometallics, 2014, 33, 2921 CrossRef CAS.
- S. Kumar, G. K. Rao, A. Kumar, M. P. Singh and A. K. Singh, Dalton Trans., 2013, 42, 16939 RSC.
- A. Kumar, M. Agarwal and A. K. Singh, Polyhedron, 2008, 27, 485 CrossRef CAS PubMed.
- A. Kumar, M. Agarwal and A. K. Singh, J. Organomet. Chem., 2008, 693, 3533 CrossRef CAS PubMed.
- A. Kumar, M. Agarwal and A. K. Singh, Inorg. Chim. Acta, 2009, 362, 3208 CrossRef CAS PubMed.
- F. Saleem, G. K. Rao, P. Singh and A. K. Singh, Organometallics, 2013, 32, 387 CrossRef CAS.
- G. K. Rao, A. Kumar, S. Kumar, U. B. Dupare and A. K. Singh, Organometallics, 2013, 32, 2452 CrossRef CAS.
- G. K. Rao, A. Kumar, B. Kumar, D. Kumar and A. K. Singh, Dalton Trans., 2012, 41, 1931 RSC.
- G. K. Rao, A. Kumar, J. Ahmed and A. K. Singh, Chem. Commun., 2010, 46, 5954 RSC.
- Q. Yao, E. P. Kinney and C. Zheng, Org. Lett., 2004, 6, 2997 CrossRef CAS PubMed.
- D. Das, G. K. Rao and A. K. Singh, Organometallics, 2009, 28, 6054 CrossRef CAS.
- E. M. Schuster, M. Botoshansky and M. Gandelman, Angew. Chem., Int. Ed., 2008, 47, 4555 CrossRef CAS PubMed.
- E. M. Schuster, M. Botoshansky and M. Gandelman, Organometallics, 2009, 28, 7001 CrossRef CAS.
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317 CrossRef CAS.
- J. Hu and Y. B. Liu, Langmuir, 2005, 21, 2121 CrossRef CAS PubMed.
- A. V. Gaikwad, A. Holuigue, M. B. Thathagar, J. E. tenElshof and G. Rothenberg, Chem.–Eur. J., 2007, 13, 6908 CrossRef CAS PubMed.
- P. J. Ellis, I. J. S. Fairlamb, S. F. J. Hackett, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2010, 49, 1820 CrossRef CAS PubMed.
- J. Rebek and F. Gaviña, J. Am. Chem. Soc., 1974, 96, 7112 CrossRef CAS.
- J. Rebek, D. Brown and S. Zimmerman, J. Am. Chem. Soc., 1975, 97, 454 CrossRef CAS.
- I. W. Davies, L. Matty, D. L. Hughes and P. J. Reider, J. Am. Chem. Soc., 2001, 123, 10139 CrossRef CAS.
- V. P. Ananikov and I. P. Beletskaya, Organometallics, 2012, 31, 1595 CrossRef CAS.
- K. J. Kilpin and J. D. Crowley, Polyhedron, 2010, 29, 3111 CrossRef CAS PubMed.
- K. J. Kilpin, E. L. Gavey, C. J. McAdam, C. B. Anderson, S. J. Lind, C. C. Keep, K. C. Gordon and J. D. Crowley, Inorg. Chem., 2011, 50, 6334 CrossRef CAS PubMed.
- C. J. Mathews, P. J. Smith and T. Welton, J. Mol. Catal. A: Chem., 2003, 206, 77 CrossRef CAS.
- M. A. Robb, J. A. Cheeseman, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, M. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, M. Nakajima, Y. Honda, K. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, V. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, J. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. A. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, G. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. A. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian-03, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed;
(b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- J. D. Webb, S. MacQuarrie, K. McEleney and C. M. Crudden, J. Catal., 2007, 252, 97 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data of L1, L2, 1 and 2; single crystal data of 1 and 2, SEM, EDX and TGA of NPs. CCDC 1013860 and 1013861. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra09574j |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.