
Monte Carlo simulation studies of cation selectivity in ion exchange of zeolites
Hiroki Nakamura*,
Masahiko Okumura and
Masahiko Machida
CCSE, Japan Atomic Energy Agency, 178-4-4 Wakashiba, Kashiwa, Chiba 277-8587, Japan. E-mail: nakamura.hiroki@jaea.go.jp
First published on 7th October 2014
Abstract
In order to evaluate the selectivity for specific cations in ion-exchange of zeolites, we calculate the ion-exchange isotherms in various zeolites using Monte Carlo simulation techniques. The calculation results agree well with experiments. Furthermore, we examine the dependence of the cation selectivity on the Si/Al ratio and find that the Cs selectivity initially increases with the Si/Al ratio but saturates above a certain value. These results reveal that the selectivity for Cs is enhanced cooperatively by the microporous structure and the Si/Al ratio of the zeolite framework. The present simulation scheme is promising in selecting useful materials for Cs decontamination in waste treatments.
1. Introduction
Zeolites are highly useful aluminosilicates employed as catalysts and adsorbents in a variety of chemical engineering processes.1 One of their most unique features is their microporous structures whose skeletal framework is formed by silicon and oxygen with partial replacement of silicon by aluminum. The microporosity throughout the crystal is selective for a particular cation, which enables the exclusive removal of the target cation from contaminated water. Their high performance has actually been proved by radioactive Cs removal from the waste water discharged in the Three Mile Island and Fukushima Dai-ichi nuclear power plant accidents. In such cases, since the highly-selective separation capacity has a key role, further performance enhancement is in great demand.The ion-exchange selectivity in zeolites is known to depend on the Si/Al ratio, although it primarily arises from the porous frame structure.2,3 For instance, mordenite and chabazite whose Si/Al ratios are relatively low belong to a high-Cs-selectivity group. Therefore, predicting the dependence of the selectivity on the Si/Al replacement ratio is a challenging target in theoretical computation. In this paper, we numerically estimate not only the cation selectivity attributed to the porous framework but also the Si/Al dependences in typical zeolites by using a semi-grand canonical Monte Carlo technique specifically developed for the ion exchange in zeolites. We reproduce their selectivity in a few known zeolites with their fixed Si/Al ratios and predict its variation depending on the Si/Al ratio in a specific zeolite.
Though various Monte Carlo techniques have been applied to silicates,4–7 Jeffroy et al.8 recently reported that their semi-grand canonical Monte Carlo simulation technique allows calculation of the ion-exchange isotherm in a zeolite. The isotherm is a typical indicator used to characterize the ion exchange performance of zeolites. In ref. 8, zeolite Y is a target in examining the ion-exchange isotherms in the presence of typical alkali metal ions. The obtained isotherms agree well with the experimental results. Following their report, we apply the technique to various zeolites to evaluate their cation selectivity. Consequently, we confirm that the technique is also applicable to different zeolites. Furthermore, we find that the Cs selectivity initially increases with increasing the Si/Al ratio but eventually saturates above a certain ratio. The technique is able to examine the coupled effects of both the porous framework structure and Si/Al ratio on the cation selectivity in a variety of zeolites.
2. Materials
We begin with the crystalline structures of zeolites. Zeolites are composed of the negatively charged aluminosilicate framework as displayed in Fig. 1, and cations trapped inside the micropores within the framework. The charge of the framework is given by the Si/Al ratio, because a replacement of Si4+ by Al3+ yields a negative charge. A trapped positive cation then compensates for the negatively charged framework. Consequently, the total number of Al atoms in the framework coincides with that of trapped univalent alkali cations. A zeolite is characterized by its porous framework structure and the Si/Al ratio in the framework.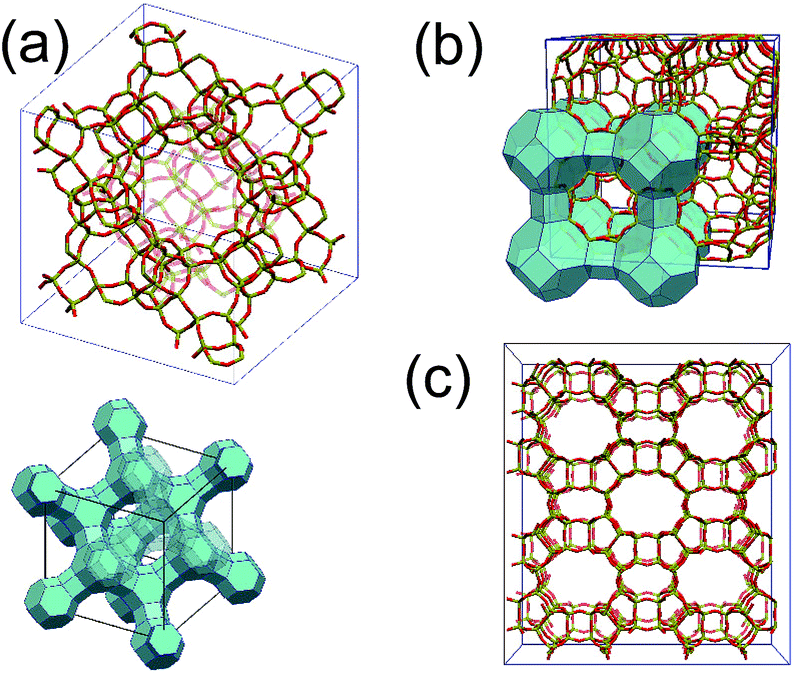 | ||
| Fig. 1 Framework structures of various zeolites, Zeolite Y (FAU) (a), Zeolite A (LTA) (b) and mordenite (MOR) (c). Dark (red) and light (yellow) bonds stand for O and Si(Al) atoms, respectively. For zeolite A and Y, schematic figures of the frameworks are also drawn. | ||
In this paper, we examine four types of zeolites, called zeolite Y, mordenite, zeolite A, and ZK-4, respectively. Zeolite Y, whose framework is faujasite-type as displayed in Fig. 1(a), has a Si/Al ratio of around 2.7. Jeffroy et al. calculated the ion exchange isotherms in zeolite Y by their semi-grand canonical Monte Carlo technique.8 We also perform the equivalent simulations in zeolite Y. We implemented their techniques in our own Monte Carlo code and confirmed validity of the code by comparing the results for zeolite Y.
Mordenite, as displayed in Fig. 1(c), is one of the useful natural zeolites employed as an efficient Cs remover in nuclear waste treatments.2 The present paper is the first report which reproduces the isotherms of mordenite to our knowledge.
Zeolite A is a famous artificial zeolite whose Si/Al ratio is around unity, belonging to a class showing the lowest value among zeolites. The selectivity for Cs in zeolite A is known to be relatively low. In the case of zeolite A, we also calculate Si/Al ratio dependent isotherms. We examine the Si/Al ratio dependence of zeolite A in an unexplored range. Note that zeolite A with a Si/Al ratio of the specific value 1.7(5/3) is called ZK-4 (ref. 9) and is distinguished from zeolite A.
3. Methods
In the present paper, we calculate isotherms of various zeolites with Si/Al ratio variations and compare the numerical results with measured ones. The isotherm comparison can demonstrate usefulness of the numerical tool in a variety of zeolites. We examine the ion exchange of Na entrapped inside zeolites with Cs or K. The inclusion of other competing ions is straightforward and their effects will be published elsewhere.Here, let us briefly illustrate the ion-exchange isotherms. For instance, we consider ion exchange of Na by Cs ion assuming the absence of other cations. We prepare a zeolite whose cation sites are filled with Na+, put it into water including Cs+ and Na+, and equilibrate the concentration of the cations in the zeolite and the solution. In this case, the ion-exchange isotherm is plotted by setting the horizontal axis as the ratio of Cs+ to total cations in the solution, XS, and the vertical axis as that in the zeolite, XZ, as shown in Fig. 2. We can easily read the Cs selectivity from the plot. If the plot is convex upward like the schematic solid curve in Fig. 2, the Cs selectivity is always higher than that of Na, because it indicates that the fraction of Cs+ in the zeolite is higher than that in the solution. On the other hand, the Cs selectivity is lower when the plot is concave downward like the dashed curve in Fig. 2.
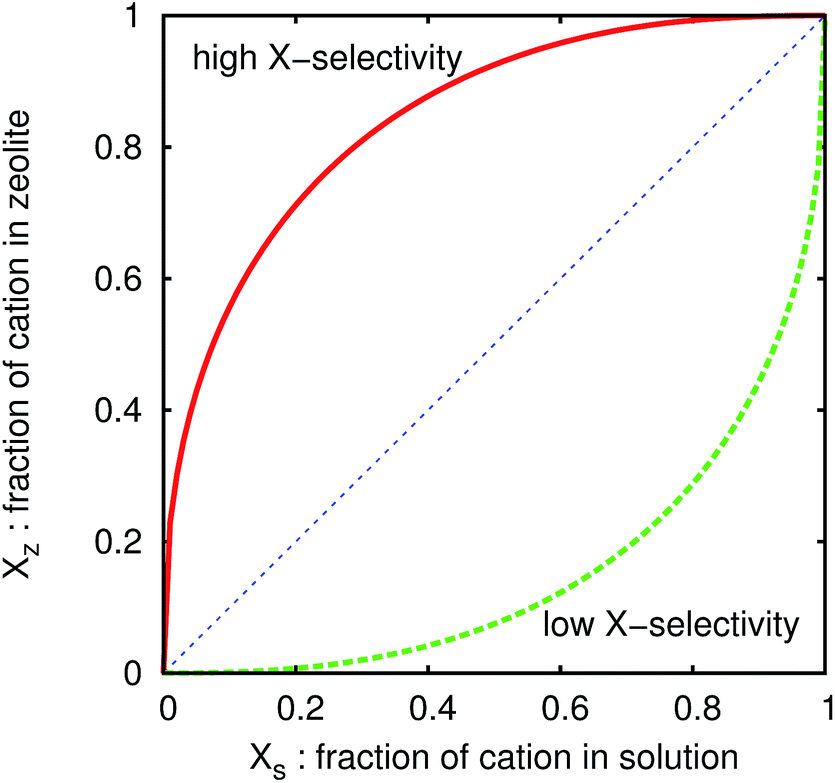 | ||
| Fig. 2 Schematic figure of an ion-exchange isotherm. The horizontal axis corresponds to the ratio of cation X to the total cations in aqueous solution, and vertical axis to that in the zeolite. If the zeolite has a high selectivity for cation X, the isotherm shows the red solid curve. In the case of low selectivity, the isotherm becomes the green dashed curve. | ||
Jeffroy et al. succeeded in reproducing ion-exchange isotherms by semi-grand canonical Monte Carlo simulation,8 which allows fractions of cations to equilibrate in solutions and zeolites. They calculated ion-exchange isotherms between Na and X (X = Li, K, Cs and Rb) in zeolite Y. In this method, “swap move” was employed in addition to usual Monte Carlo moves such as translations and rotations (see e.g., ref. 10). In the swap move, the species of cations are randomly changed with keeping the total number of cations fixed, and new cation coordination is accepted or rejected by the Metropolis method for fixed chemical potential difference between two cations. This has a key role in describing the ion-exchange process.
In the present simulation, the Lennard-Jones and Coulomb potential are employed as the force fields. The form of the Lennard-Jones potential between the i- and j-th atoms is expressed as −4εij[σij6/r6 − σij12/r12] with the Lorentz–Berthelot combination rules, σij = (σi + σj)/2 and  . The parameters of the potential are summarized in Table 1. The Ewald summation is used for the long-range Coulomb interactions.13 The water model in our calculation is TIP4P.14 With these force fields, Na–K and Na–Cs ion exchanges are examined. For the initial aqueous solution, we prepare 0.1 M NaCl solution by the system of 2000 water molecules with 4Na+ and 4Cl− ions in the cubic cell, 39.15 Å on a side, and equilibrate the ratios of Na and X (X = K and Cs) to total cations by Monte Carlo simulation for the fixed chemical potential difference between Na and X. Next, a similar type of simulation is performed for zeolites together with cations and water molecules, and the ratio of X to total cations in zeolites is then obtained. The cell size and the number of atoms in each simulation are summarized in Table 2, and all the framework structures of the calculated zeolites are taken from ref. 11 and 15. Finally, the ion-exchange isotherms are obtained by plotting the ratios of X in the zeolite and the solution at various chemical potential differences.
. The parameters of the potential are summarized in Table 1. The Ewald summation is used for the long-range Coulomb interactions.13 The water model in our calculation is TIP4P.14 With these force fields, Na–K and Na–Cs ion exchanges are examined. For the initial aqueous solution, we prepare 0.1 M NaCl solution by the system of 2000 water molecules with 4Na+ and 4Cl− ions in the cubic cell, 39.15 Å on a side, and equilibrate the ratios of Na and X (X = K and Cs) to total cations by Monte Carlo simulation for the fixed chemical potential difference between Na and X. Next, a similar type of simulation is performed for zeolites together with cations and water molecules, and the ratio of X to total cations in zeolites is then obtained. The cell size and the number of atoms in each simulation are summarized in Table 2, and all the framework structures of the calculated zeolites are taken from ref. 11 and 15. Finally, the ion-exchange isotherms are obtained by plotting the ratios of X in the zeolite and the solution at various chemical potential differences.
| Lennard-Jones | Charge | ||
|---|---|---|---|
| σ (Å) | ε (K) | q (e) | |
| OZ | 3.00 | 93.53 | — |
| SiZ(AlZ) | 3.302 | 0.001 | — |
| Na | 2.584 | 50.27 | 1.0 |
| K | 2.907 | 62.00 | 1.0 |
| Cs | 3.165 | 59.72 | 1.0 |
| Cl | 4.450 | 50.28 | −1.0 |
| OW | 3.1536 | 78.03 | 0.0 |
| HW | 0.0 | 0.0 | 0.52 |
| XW | 0.0 | 0.0 | −1.04 |
| Name | Chemical formula | Framework type | Lattice parameters | Si/Al ratio | Selectivity | Charge | |||
|---|---|---|---|---|---|---|---|---|---|
| a (Å) | b (Å) | c (Å) | OZ | SiZ(AlZ) | |||||
| a At low loading. | |||||||||
| Zeolite Y | Na52Al52Si140O384·250H2O | FAU | 24.345 | 24.345 | 24.345 | 2.6923 | Cs > K > Naa | −0.8245 | 1.3781 |
| Mordenite | Na64Al64Si320O768·192H2O | MOR | 36.512 | 41.068 | 15.084 | 5 | Cs > K > Na | −0.8140 | 1.4613 |
| Zeolite A | Na96Al96Si96O384·216H2O | LTA | 23.838 | 23.838 | 23.838 | 1 | Na > K > Cs | −0.8500 | 1.2000 |
| ZK-4 | Na72Al72Si120O384·216H2O | LTA | 23.838 | 23.838 | 23.838 | 1.6666 | Cs > Na | −0.8365 | 1.2980 |
| Na48Al48Si144O384·216H2O | LTA | 23.838 | 23.838 | 23.838 | 3 | — | −0.8230 | 1.3960 | |
| Na24Al24Si168O384·216H2O | LTA | 23.838 | 23.838 | 23.838 | 7 | — | −0.8095 | 1.4940 | |
In these Monte Carlo simulations, we adopt translational and rotational moves for water molecules, and translational moves for ions. In addition, swap move is applied to exchangeable cations. The coordinates of Si(Al) and O atoms in the zeolite frameworks are fixed through simulations. We check equilibration by convergence of total energy. In the simulations of aqueous solution and zeolites, energy convergences are observed after 1.5 × 107 and 1.0 × 107 steps, respectively. Statistical averages are taken with respect to 1.5 × 107 and 4 × 107 steps after equilibration in the cases of aqueous solution and zeolite, respectively. To increase accuracy, we start simulations from ten different randomly-distributed initial coordinates of water and ions. We plot the statistical errors in Fig. 3(b). Though the errors of the cation ratio in solution are slightly large due to lack of sampling, it is accurate enough to examine the selectivity. All the calculations are executed by our in-house code, while the validity of our code is confirmed by comparing with MedeA-GIBBS (commercial code) implementing the semi-grand Monte Carlo technique.16
 | ||
| Fig. 3 Ion-exchange isotherms for Na–Cs ion exchanges in zeolite Y (a), mordenite (c), zeolite A (e) and ZK-4 (g), and for Na–K ion exchange in zeolite Y (b), mordenite (d), zeolite A (f) and ZK-4 (h). The horizontal axis corresponds to the fraction of cation X (X = Cs, K) to the total cations (X and Na) in solution, and the vertical axis to that in zeolite. Filled circles and squares denote experimental data, while solid, dashed and dotted curves show calculated results. In (b), the error bars denote the statistical errors. In (g) and (h), the results with Si/Al ratios of 5/3, 3 and 7 correspond to solid, dashed and dotted curves, respectively. Experimental data are taken from ref. 17–20. | ||
We note that the interactions between cations and oxygen atoms in zeolite frameworks are different from those in ref. 8. While the Buckingham potential is employed in ref. 8, the Lennard-Jones potential is additionally tested in the present work. We mention that the isotherms of zeolite Y (Fig. 3(a) and (b)) calculated by the Lennard-Jones potential also agree well with experiments. We point out that the Lennard-Jones type is also valid in various zeolites. While more accurate and complicated potentials have been developed, we believe that Coulomb and Lennard-Jones potentials are adequate in this Monte Carlo simulation. Since neither chemical reaction nor charge transfer occur in the ion exchange process, interactions among water molecules, ions and zeolite frameworks can be described well by Coulomb and Lennard-Jones potentials.
4. Results
In order to check whether the present Monte Carlo technique can be applied to other zeolites, we evaluate Na–K and Na–Cs ion-exchange isotherms for mordenite and zeolite A. Mordenite has a relatively high Si/Al ratio and shows much higher Cs selectivity than that of zeolite Y. See Fig. 1(c) for its framework structure. The electric charges of atoms in the mordenite framework are determined from the Si/Al ratio according to ref. 12 and are shown in Table 2. Fig. 3(c) and (d) show the calculated isotherms. They agree very well with the observed ones. These results indicate that the present numerical technique actually reproduces the very high Cs and K selectivity of mordenite as measured in experiments. In Fig. 4(a), we also show the Cs sites in mordenite obtained by this simulation. | ||
| Fig. 4 Cs density in the unit cells of mordenite (a) and zeolite A (b). Blue dots show the density of Cs ions. Yellow and red cylinders denote bonds from Si and O atoms of the zeolites, respectively. | ||
Zeolite A is known to have lower Cs selectivity. The calculated isotherms for zeolite A are shown in Fig. 3(e) and (f), and calculated Cs sites are shown in Fig. 4(b). In both the Na–K and the Na–Cs ion exchanges, our results for zeolite A are slightly lower than the observed ones but almost consistent with the experiments. Thus, the present Monte Carlo method is suitable for numerically calculating the isotherms.
As mentioned above, the Cs selectivity is generally known to be improved when the Si/Al ratio is increased. However, it is a rather naive view that elevating the Si/Al ratio always improves Cs selectivity, since the role may be limited in selectivity enhancement. To clarify the effect of the Si/Al ratio, we calculate isotherms for zeolites with Si/Al ratios of 5/3, 3 and 7 with the same framework as zeolite A. In these calculations, the variation of the Si/Al ratio is realized by controlling the electric charges of atoms in the zeolite framework. According to ref. 12, we set the electric charges as shown in Table 2. The calculated results for the different Si/Al ratios are shown in Fig. 3(g) and (h). From these figures, the Cs and K selectivity actually increases with increasing the Si/Al ratio. However, the isotherms for Si/Al ratios of 3 and 7 are almost the same. Thus, we find that the Cs and K selectivity increases with the Si/Al ratio while it saturates above a certain value.
5. Discussion
The dependence of the cation selectivity on the Si/Al-ratio can be explained as follows.21,22 For smaller cations such as Na, the relative distances between the cations and the given framework are so short that the interaction energy between them is sensitive to the charge variation of the framework since the Coulomb energy is proportional to inverse of those distances. As such, the Na ions are affected more strongly by the change in the Si/Al ratio than the larger Cs and K ions. Namely, the Na interaction energy increases with the Si/Al ratio, and the K and Cs selectivity is relatively less enhanced.The next topic is the reason why the selectivity saturates above a certain high Si/Al ratio. The largest Si/Al ratio in zeolites employed in this paper is 7 for the ZK-4-type zeolite. In the case of mordenite, its Si/Al ratio is 5 while it shows the highest Cs selectivity as shown in Fig. 3(c). Therefore, the Cs selectivity is not controlled only by the Si/Al ratio. Rather, other factors such as framework structure are also found to affect the selectivity strongly. In the ZK-4 or zeolite A, cations are mainly absorbed in 8-membered and 6-membered rings (8 MR and 6 MR) composed of oxygen atoms in zeolite frameworks.23 These rings are almost flat and two-dimensional. According to the ion radii, 6 MR fit Na, and 8 MR are large enough for both Na and Cs. When the Si/Al ratio increases, the number of cations decreases, and then most of the Na ions are captured by 6 MR. The Cs selectivity enhancement by the high Si/Al ratio is canceled by the capture of Na inside 6 MR. This can be a reason for the saturation. On the other hand, most of the cation sites in mordenite are composed of 8 MR which are distorted and three-dimensional. These distorted 8 MR are more suitable for Cs absorption compared to the flat 8 MR of zeolite A. Consequently, the mordenite framework shows very high Cs selectivity. Thus, the framework structure also plays an important role for the cation selectivity.
6. Conclusions
In conclusion, in order to numerically evaluate the cation selectivity in ion exchange, we calculated the ion-exchange isotherms for zeolite Y, zeolite A, mordenite, and ZK-4, using the Monte Carlo simulation technique developed in ref. 8. The obtained calculation results are almost consistent with experiments. We also examined the dependence of the cation selectivity on the Si/Al ratio. Consequently, we found that the Cs and K selectivity increases with the Si/Al ratio and saturates above a certain high Si/Al ratio as seen in the case of the zeolite-A framework. We conclude that high Cs selectivity arises from the cooperative effects between the framework structure and the Si/Al ratio and believe that this conclusion is valid in the cases of other zeolites which are not investigated in this paper. Finally, we point out that the present calculation tool is useful in exploring zeolites with high selectivity for radioisotope ions. It may open a new research pathway for nuclear waste management.Acknowledgements
We would like to thank I. Yamagishi, Y. Koma, A. Shibata, Y. Goto and K. Kobayashi for their helpful discussions. All the calculations were performed on BX900 in CCSE, JAEA. We thank the director of CCSE, M. Tani, and all the other staff members of BX900. In addition, we thank A. Fujiwara, K. Mori and staffs of Materials Design Inc. for their technical advice. This work is partially supported by the JAEA-NIMS Collaborative Fukushima Recovery Project organized by T. Yaita. We thank all the project members for their illuminating discussions.References
- S. Auerbach, K. Carrado and P. Dutta, Handbook of Zeolite Science and Technology, Marcel Dekker, New York, 2003 Search PubMed.
- A. Dyer, in Introduction to Zeolite Science and Practice, ed. J. Čejka, H. van Bekkum, A. Corma and F. Schüth, Elsevier, Amsterdam, 3rd revised edn, 2007, ch. 16, p. 525 Search PubMed.
- H. S. Sherry, in Handbook of Zeolite Science and Technology, ed. S. Auerbach, K. Carrado and P. Dutta, Marcel Dekker, New York, 2003, ch. 21, p. 1007 Search PubMed.
- B. Smit, Mol. Phys., 1995, 85, 153–172 CrossRef CAS.
- S. Pu, Y. Tanaka and T. Inui, Sep. Technol., 1996, 6, 189–195 CrossRef CAS.
- X.-Q. Zhang, T. T. Trinh, R. A. van Santen and A. P. J. Jansen, J. Am. Chem. Soc., 2011, 133, 6613–6625 CrossRef CAS PubMed.
- X.-Q. Zhang, R. A. van Santen and A. P. J. Jansen, Phys. Chem. Chem. Phys., 2012, 14, 11969–11973 RSC.
- M. Jeffroy, A. Boutin and A. H. Fuchs, J. Phys. Chem. B, 2011, 115, 15059–15066 CrossRef CAS PubMed.
- G. T. Kerr, Inorg. Chem., 1966, 5, 1537–1539 CrossRef CAS.
- J. M. Thijssen, Computational Physics, Cambridge, 1999 Search PubMed.
- C. Baerlocher, L. B. McCusker and D. H. Olson, Atlas of Zeolite Framework Types, Elsevier, Amsterdam, 2007 Search PubMed.
- A. Di Lella, N. Desbiens, A. Boutin, I. Demachy, P. Ungerer, J.-P. Bellat and A. H. Fuchs, Phys. Chem. Chem. Phys., 2006, 8, 5396–5406 RSC.
- A. Y. Toukmaji and J. A. Board Jr, Comput. Phys. Commun., 1996, 95, 73–92 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS PubMed.
- C. Baerlocher and L. B. McCusker, Database of Zeolite Structures, http://www.iza-structure.org/databases/ Search PubMed.
- MedeA-Gibbs licensed from: IFP Energies nouvelles and Laboratoire de Chimie-Physique, Materials Design, Inc., Université de Paris-Sud, CNRS; MedeA version 2.13, Angel Fire, NM, USA, 2013 Search PubMed.
- H. S. Sherry, J. Phys. Chem., 1966, 70, 1158–1168 CrossRef CAS.
- F. Wolf, H. Fürtig and H. Knoll, Chem. Techn., 1971, 23, 273 CAS.
- L. L. Ames, Can. Mineral., 1965, 8, 325–333 CAS.
- R. M. Barrer, L. V. C. Rees and D. J. Ward, Proc. R. Soc. London, Ser. A, 1963, 273, 180–197 CrossRef.
- H. Nakamura, M. Okumura and M. Machida, J. Phys. Soc. Jpn., 2013, 82, 023801 CrossRef.
- G. Eisenman, Biophys. J., 1962, 2, 259 CrossRef CAS.
- W. J. Mortier, Compilation of Extra Framework Sites in Zeolites, Butterworth, Guildford, 1982 Search PubMed.
| This journal is © The Royal Society of Chemistry 2014 |