
Designing triphenylamine derivative dyes for highly effective dye-sensitized solar cells with near-infrared light harvesting up to 1100 nm†
Abstract
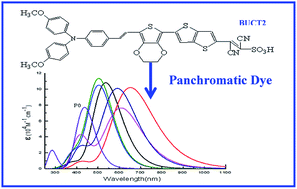
Designing highly efficient sensitizers for dye-sensitized solar cells (DSSCs) is an urgent task because it is closely related to the practical application of DSSCs. In this work, we designed and screened a series of triphenylamine derivative dyes with donor–π–acceptor (D–π–A) structure using different electron donors, π bridges and electron acceptors, and further used density functional theory (DFT) and time-dependent DFT (TDDFT) approaches to investigate the molecular orbital energy levels, absorption spectra, and light harvesting efficiency of these newly designed dyes. Results indicate that the donor group in D2, π bridges in Pi10–Pi12 and the acceptor group in A7 are promising functional groups for D–π–A structure. Using the above screened functional groups as donors, π bridges and acceptors, we designed six novel D–π–A structures of BUCT1–BUCT6. The results indicate that BUCT1–BUCT6 dyes show smaller HOMO–LUMO energy gaps, higher molar extinction coefficients and obvious redshifts compared to the experimentally synthesized P0 dye. In particular, the newly designed BUCT2 dye exhibits not only a 215 nm redshift and a higher molar extinction coefficient with an increment of 32.4% compared to P0 dye, but also has an extremely broad absorption spectrum covering the entire visible range up to the near-IR region of 1100 nm. Therefore, the BUCT2 dye is a very promising candidate for highly effective DSSCs with near-infrared light harvesting up to 1100 nm. We also found that the dyes with two –CN groups and a sulfonic acid group as the electron acceptor are more efficient than dyes with one –CN group and a sulfonic acid group.
 Please wait while we load your content...
Please wait while we load your content...

