
Bottom-up approach to engineer a molybdenum-doped covalent-organic framework catalyst for selective oxidation reaction†
Abstract
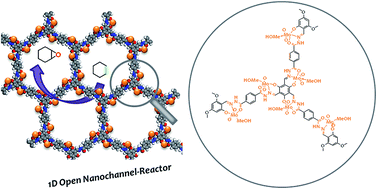
We synthesized a readily accessible molybdenum-doped covalent-organic framework catalyst (Mo-COF) linked by a hydrazine linkage via a facile two-step bottom-up approach. This Mo-COF catalyst, as an open nanochannel-reactor, exhibited promising catalytic activity for the selective oxidation reaction.
 Please wait while we load your content...
Please wait while we load your content...

