
Reusable colloidal graphene oxide suspensions combined with dialysis bags for recovery of trace Y(iii) from aqueous solutions†
Abstract
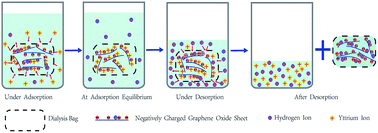
Recycling trace rare earth elements (REEs) from effluents from mines and refineries is vital for the protection of the global environment and utilization of valuable REE resources. In this work, a novel and highly efficient method for second-pollution-free adsorptive recovery of trace Y(III) from aqueous solutions by colloidal graphene oxide (GO) suspensions loaded in dialysis bags has been developed based on the properties of GO suspensions and the sieving characteristics of dialysis membranes. The effects of pH, ionic strength and temperature on Y(III) adsorption–desorption on GO were studied. Thermodynamic and kinetic investigations on Y(III) adsorption on GO were also conducted. The maximum adsorption capacity of Y(III) on GO at pH = 5.9 ± 0.1 and T = 303 K was 190.48 mg g−1, higher than any other adsorbents reported so far. In the fourth consecutive adsorption–desorption cycle, the Y(III)-saturated GO suspension was still able to resume a colloidal state in pH = 0.9 HNO3 aqueous solution with a desorption rate of 74.26% while the regenerated GO maintained an adsorption capacity of 138.73 mg g−1. The adsorption–desorption capacities of Y(III) on GO depended more on pH than on ionic strength and temperature. The studies on adsorption thermodynamics and kinetics showed that the adsorption of Y(III) on GO was an endothermic, spontaneous and monolayer adsorption process, and perfectly fitted the Langmuir model and the pseudo-second-order model. In summary, GO is a highly efficient and easily reusable adsorbent with promising application for the treatment of industrial wastewater and the enrichment and recovery of lanthanides. More importantly, the combination of colloidal GO suspension with dialysis membrane facilely avoids the re-pollution of the treated solutions, drastically reduces workload in separation and recovery of GO and provides an alternative route for actual application of nano-sized adsorbents in environmental pollutant removal and valuable resource recovery from wastewater.
 Please wait while we load your content...
Please wait while we load your content...

