
Improvement of CdSe quantum dot sensitized solar cells by surface modification of Cu2S nanocrystal counter electrodes
Abstract
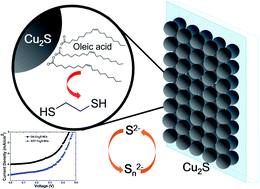
We report the improvement of a CdSe quantum-dot-sensitized solar cell (QDSSCs) based on surface modification of Cu2S nanoparticle counter-electrodes (CEs). In this work, we explored a low-cost, easy method to fabricate counter electrodes by direct deposition of colloidal Cu2S NCs on conducting FTO glass using drop casting or spin coating. The colloidal Cu2S NC films provide high surface area, which improves the catalytic activity for the redox couple and enhances the final photovoltaic performance. A CdSe QDSSC based on the 0.1 M EDT treated Cu2S CE/FTO shows a short-circuit current density (JSC) of 7.95 mA cm−2, a fill factor (FF) of 55.44%, and yielded a superior power conversion efficiency (η) of 2.1%, an improvement of 50% over that of the OA-capped Cu2S CE/FTO CE (1.4%).
 Please wait while we load your content...
Please wait while we load your content...

