
Liquid–liquid phase separation and its effect on the crystallization in polylactic acid/poly(ethylene glycol) blends
Abstract
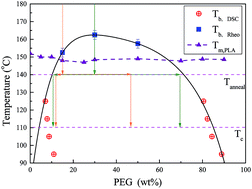
The miscibility in blends of polylactic acid (PLA) and poly(ethylene glycol) (PEG) was investigated by means of differential scanning calorimetry (DSC), polarized optical microscopy (POM) and rheology using a specifically designed thermal procedure. The direct observation of a phase separated morphology using phase contrast optical microscopy, is difficult because of the quite close refractive indices of the two components. Liquid–liquid phase separation (LLPS) was confirmed by the appearance of two glass transitions after the samples have been annealed at low temperatures (95–125 °C). Moreover, the accelerated crystallization rate after the sample annealing at high temperatures (140–160 °C) also indicated the existence of LLPS, according to a fluctuation-assisted nucleation mechanism. An upper critical solution temperature (UCST) type of phase diagram was derived using the combination of DSC and rheological methods. Furthermore, the effects of LLPS on the kinetics of the subsequent crystallization of PLA in blends with 15 wt% PEG and 30 wt% PEG were investigated. Considering the complex effects of LLPS on the crystallization, we proposed a new model that integrates the Lauritzen–Hoffman theory and the self-concentration theory to account for the influence of miscibility on the crystallization in blends with different thermal histories. It is suggested that the effect of acceleration or deceleration of LLPS on the subsequent crystallization in a highly asymmetric system, depends on the interplay between the fluctuation-assisted nucleation and the variation of mobility during LLPS.
 Please wait while we load your content...
Please wait while we load your content...

