
A facile one-pot route towards three-dimensional graphene-based microporous N-doped carbon composites†
Zehui Zhang and
Peiyi Wu*
State Key Laboratory of Molecular Engineering of Polymers and Department of Macromolecular Science and Laboratory of Advanced Materials, Fudan University, Shanghai 200433, P. R. China. E-mail: peiyiwu@fudan.edu.cn
First published on 15th September 2014
Abstract
3D graphene aerogel-based microporous N-doped carbon composites can be prepared by hydrothermal assembly of graphene oxide with chitosan. A large amount of chitosan added into the system acts as a coating to adhere to the graphene sheets and separate the graphene. During the hydrothermal reaction, the GO is reduced and forms 3D graphene-based frameworks which provide the 3D template for the efficient growth of N-doped carbon on the graphene surface, and the chitosan, which went through a dehydration and carbonization process, forms N-doped carbon strongly adhered to the graphene sheets due to the π–π stacking interactions which effectively solve the aggregation problems between the graphene layers. Furthermore, the resulting microporous N-doped carbon composites show enhanced supercapacitive performance and high efficiency as metal-free catalysts for oxygen reduction reaction.
Introduction
Graphene, because of its one atom thickness, large aspect ratio, high surface area, good electrical conductivity and many interesting physical properties, is thought to be a promising material in various fields.1–6 Nowadays, three-dimensional (3D) graphene-based frameworks, such as aerogels,7,8 foams,9,10 and sponges,11,12 have attracted much attention for their continuously interconnected macroporous structures, low mass density, large surface area, and high electrical conductivity, and also have been served as template for accommodating metal, metal oxide, and electrochemically active polymers for various applications in electrochemical capacitors, catalysis and other applications.13–16 But these materials always lack well-defined pores, which greatly limits their use in many applications, especially the efficiency of the transport and charge storage in electrochemical capacitors.17To date, many approaches have been used to prepare graphene-based hybrid materials with hierarchical porous structures.17–28 Most of these approaches rely on the use of graphene-based porous silica template17,18 with many kinds of nanoparticles decorated such as metal oxide19–21 and metal sulfide.22–24 Conjugated microporous polymer sheets25 and mesoporous carbon26–28 were also used to prepare porous graphene nanohybrid. But these methods always need tedious preparation processes and/or not environmental friendly. Then, a facile method to prepare graphene based hybrid with micropores is highly attractive.
Recently, many researchers have found that carbohydrate can be used to fabricate graphene aerogel,29,30 but how these carbohydrates help fabricate graphene aerogel and what these carbohydrates really go in the end have not been revealed yet. Herein, we report a facile graphene-inspired synthetic approach to the large-scale production of 3D graphene aerogels based micropores N-doped carbon composites (GA–NC) in which graphene sheets can be effectively separated by micropores carbon shell. The resulting GA–NC can be prepared by hydrothermal assembly of graphene oxide (GO), while the N-doped carbon can be in situ grown on the graphene surface at the same time by the hydrothermal carbonization approach. Different as the previously reports which indicated that the chitosan act as spacer and reduction during the hydrothermal reaction,29,30 we found that large amount of chitosan added into the system act as coatings to adhere on the graphene sheets and separate the graphene. The aggregation of graphene oxide (GO) during the hydrothermal reaction can be effectively suppressed by the micropores carbon layers (coatings) and would not aggregate easily. The GA–NC possesses high surface areas and large aspect ratios. These unique structural features associated with graphene network confined within the N doped micropores carbon establish a new generation of porous materials with multiple functions.
Experimental
Chemicals
Graphite powders, chitosan, KOH were purchased from Sinopharm Chemical Reagent Co., Ltd (China). All chemicals from commercial sources were used as received without further purification. All aqueous solutions were prepared with Milli-Q water (18.2 MΩ cm−1).Synthesis of GA–NC
Graphite oxide powders were synthesized from graphite powders using a modified Hummers method. The GO (400 mg) was dissolved in water with the concentrations of 2.5 mg mL−1. The GO/H2O solutions were under ultrasonication for 6 h.Typically, 10 mL GO/H2O solutions was put into a 25 mL Teflon-lined autoclave, and different amount of chitosan (1.5 g, 1 g, 0.5 g) was added into the autoclave. Afterwards, the autoclave was heated at 220 °C for 12 h and gradually cooled down to room temperature. Followed by drying at 80 °C and annealing at 800 °C for 3 h in N2 gas. The GA–NC hybrids prepared with different amount of chitosan were denoted as GA–NC-1.5, GA–NC-1, GA–NC-0.5. The pristine GA and pristine N-doped carbon composites (NC) was prepared with the same process here for comparison.
Electrochemical measurements
The Electrochemical experiments for supercapacitor were carried out in a standard three-electrode system, which contained a nickel foil electrode as a counter electrode, an Hg/HgCl2 as a reference electrode, GNSs loaded on foam nickel as a working electrode and KOH aqueous solution (6 mol L−1) as electrolyte. All electrochemical measurements were carried out on a CHI 660B electrochemical working station. Galvanostatic charge–discharge test was taken within the voltage range of −1 to 0 V, which was used to calculate the specific capacitance of GA–NC electrodes from the slope of discharge curves (dV/dt). For the cyclic voltammetric measurements, the sweep rate ranged from 5 to 200 mV s−1 within a potential range of −0.8 to 0.2 V. For the EIS measurements, the frequency range was from 1 Hz to 10 kHz.The Electrochemical experiments for ORR were carried out in a standard three-electrode system, (CHI660D) in a standard three-electrode cell using Ag/AgCl electrode as the reference electrode, a platinum wire as the counter electrode and a glassy carbon electrode as the working electrode. The preparation of working electrode was referred to our previous work. In brief, 5 mg catalyst was dispersed in 3 mL deionized water by 45 min sonication to form homogeneous suspension. Then 5 μL suspension was dropped onto a glassy carbon electrode with 3 mm diameter, and 5 μL Nafion solution (5 wt%) was coated after the suspensions was dried. The three electrodes were put into 0.1 M KOH solution. A flow of O2 or N2 was maintained for 40 min to achieve O2-saturated or O2-free. The working electrode was cycled at least 5 times in the potential range from −0.8 V to 0.2 V at a scanning rate of 5 mV s−1.
Characterization
Transmission electron microscopy (TEM) experiments were conducted on a JEOL JEM-2100F microscope (Japan) operated at 200 kV. The samples for the TEM measurements were supported onto a holey carbon film on a Cu grid. Scanning electron microscopy (SEM) experiments were conducted on a FE-SEM S-4800 (Japan). Fourier transform infrared (FTIR) spectra were recorded on a Nicolet Nexus 470 spectrometer using spectroscopic grade KBr. Raman spectra were recorded on a Renishaw inVia Reflex (514 nm). The isotherm plots were recorded on an ASAP 2020 – Physisorption Analyzer (America). The C, H and N contents were measured on a Vario EL III elemental analyzer (Germany). X-ray photoelectron spectroscopy (XPS) experiments were carried out on a RBD upgraded PHI-5000C ESCA system (Perkin Elmer) with Mg Kα radiation (hν = 1253.6 eV), with a chamber pressure of 1 × 10−8 Torr.Results and discussion
As shown in Scheme 1, GA–NC in this study was fabricated by in situ hydrothermal self-assembly reaction, followed by an annealing process. Firstly, graphene oxide sheets (GO) were mixed with chitosan in water to form a turbid liquid. Then the hydrothermal treatment of the obtained turbid liquid leads to a black aerogel (Fig. S1†). Followed by drying and annealing in N2 gas, GA–NC was successfully constructed. During these process, the GO constructs a three-dimensional (3D) graphene-based framework which provides 3D template for the growth of N-doped carbon (NC), and the chitosan, which went through a dehydration and carbonization process,31–33 forms NC strongly adhered on the graphene sheets which effectively solves the aggregation problems between the graphene layers. After heat annealing, the NC forms micropores carbon wall on the graphene-based frameworks and GA–NC formed. The pristine GA and NC nanoparticle were also prepared for comparison in this work. | ||
| Scheme 1 Schematic illustration of the synthesis of GA–NC. | ||
The morphology and microstructure of the samples were investigated by means of scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in Fig. 1a, pristine NC shows carbonized spherical colloidal particles which are similar as that reported before.32,33 As the GO added into the system, the samples showed completely different morphology. As shown in Fig. 1c, e and f, the GA–NC showed a 3D interconnected frameworks which were similar as the pristine GA (Fig. 1b), which means that a small amount of GO added can greatly change the morphology of NC during the hydrothermal process. When a small amount of GO was added into the system, only a small amount of free NC spheres were appeared which were adhered on the RGO sheets and the RGO sheets were effectively separated and formed a 3D frameworks (Fig. 1c). With the GO further increasing, free NC spheres disappeared and the composites form a 3D interconnected frameworks which was similar as GA (Fig. 1e). With the GO continuing increasing, part of the RGO sheets aggregated (Fig. 1g). The TEM results further reveal the structure of the composites. As shown in Fig. 1d, f and h, the graphene like sheets were NC coated RGO sheets in a sandwich-like manner with a typical thickness of 10 nm. Different as the silicon based GA,17 the thickness of the NC wall here is restricted and cannot be tune, the excess NC always formed free NC spheres which is similar as the pristine NC. Obviously, for GA–NC-1, most NCs formed during the hydrothermal reaction have been homogeneously adhered on the surface of graphene with no free NCs sphere and no aggregate RGO sheets. These results strongly suggest the crucial role of graphene as a substrate for the NCs in a 2D manner.
 | ||
| Fig. 1 Morphology and microstructure of samples. (a and b) Low magnification SEM images of pristine NC and pristine GA sample, inset: high-magnification SEM images of the correspond samples; (c and d) representative SEM (inset: high-magnification SEM images) and TEM images of GA–NC-1.5; (e and f) representative SEM (inset: high-magnification SEM images of GA–NC-1) and HRTEM images of GA–NC-1; (g and h) representative SEM (inset: high-magnification SEM images of GA–NC-0.5) and HRTEM images of GA–NC-0.5. | ||
The typical TEM images further disclose the porous and amorphous nature of the composites27 which is similar as the hydrothermal carbonization of diverse biomass and biomass derivatives.31 The adsorption–desorption curve exhibits the prominent characteristic of type-IV isotherms with a distinct hysteresis loop of H2, implying the presence of micropores in the frameworks (Fig. S2†). Brunauer–Emmett–Teller (BET) areas of GA–NC composites reveals of higher specific surface area than that of GA and NC. The graphene here can not only offer a template for the NC, but also be effectively separated by the NC adhered. Obviously, the GA–NC-1 possess the highest specific surface area of 353.7 m2 g−1. Interestingly, a well-defined 1.2 nm micropore was obtained (Fig. S3†) for the micropores NC wall adhered on the graphene. These results indicate that the high surface area of composites with micropores can be achieved by taking advantage of the 3D framework of GAs with NC wall adhered. The NC wall here can not only offer micropores, but also restrict the aggregation of RGO sheets during the hydrothermal process.
As the NCs uniformly grows on both sides of RGO in a sandwich-like manner and the typical thickness of N-doped carbon walls is around 10 nm, a π–π stacking interactions inspired forming mechanism was proposed here to explain the formation process. During the hydrothermal reaction, the chitosan formed N-doped cross-linked aromatic clusters which can form strong π–π stacking interactions with RGO sheets as shown in Scheme 2. The aromatic clusters of NC grew thick and formed uniform walls on the RGO sheets. However, the thickness of NC walls cannot grow infinitely and is limited by the π–π stacking interactions between the aromatic clusters of NC and the RGO sheets. With the NC wall thickening, the π–π stacking interactions weakened and the outmost aromatic clusters cannot be restricted and formed free NC spheres adhered. Because the NC can form micropores after the heat annealing, we finally got a micropores NC coated 3D-graphene framework.
 | ||
| Scheme 2 Schematic illustration of the strong π–π stacking interactions between the aromatic clusters of NC and the RGO sheets. | ||
For the highest surface area GA–NC-1 possessing, its chemical nature was further investigated by X-ray photoelectron spectroscopy (XPS), Energy Dispersive Spectrometer (EDS) and elemental analysis. The XPS spectra (Fig. 2a) display binding energy peaks at 285, 400, and 532 eV, which can be attributed to the C1S, N1S, and O1S photoelectric lines, respectively. The spectrum of C1S centered at 285 eV (Fig. 2b) is commonly observed for CN materials because of the dominant existence of C–N bonds in the matrix, and it can be deconvoluted into several single peaks that correspond to C–C (284.5 eV), C–N (285.9 eV), C–O (286.5 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (287.8 eV) and C(O)O (287.8 eV) functional groups. The complex N1S spectrum can be further deconvoluted into two different signals with binding energies of 398.0, and 401.3 eV that correspond to pyridinic N (N1) and graphitic N (N3) respectively. The EDS result indicates that the N elements dispersed uniformly on the graphene sheets which further reveal that the NC homogeneously attached on the surface of graphene to form sandwich-like composites which is in agreement with the XPS result (Fig. S4†). The elemental analysis results (Table S1†) further reveal that the composite is composed of C (74.65%), H (1.67%), N (6.30%), and O (calculated, 17.38%) atoms, which is consistent with the EDX results. All these results showed that the GA–NC-1 is high content of N doped graphitization carbon composites. Raman spectroscopy was used to confirm the quality of the GA–NC-1 (Fig. S5†). Two major features, a D band and G band, were observed at around 1385 and 1575 cm−1 respectively. The relative intensity of the “disorder” D-band and the crystalline G-band (ID/IG) for the GA–NC-1 is about 1.1, indicating its disorder and amorphous feature. The FTIR spectrum of GA–NC-1 was shown in Fig. S6.† Corresponds to the previous XPS result, the peak at 1121 cm−1 is caused by the vibration of C–O, covers the characteristic peak of graphite. The shoulder peak around 1542 cm−1 reflects the skeletal vibration of graphene and the aromatic clusters of NC. The peaks around 2925 cm−1, which corresponds to the C–H stretching mode, suggest that the NC may be broken into small pieces with more edges which also correspond to the amorphous nature of GA–NC-1. Therefore, we can conclude that the as-prepared GA–NC-1 is made up of the graphene sheets coated with N-doped amorphous carbon which is mainly composed of pyridinic N and graphitic CN species derived from the dehydration and carbonization of chitosan. Thus, this strategy provides a feasible way to build up 3D porous carbons decorated graphene composites with a high level of N doping.
O (287.8 eV) and C(O)O (287.8 eV) functional groups. The complex N1S spectrum can be further deconvoluted into two different signals with binding energies of 398.0, and 401.3 eV that correspond to pyridinic N (N1) and graphitic N (N3) respectively. The EDS result indicates that the N elements dispersed uniformly on the graphene sheets which further reveal that the NC homogeneously attached on the surface of graphene to form sandwich-like composites which is in agreement with the XPS result (Fig. S4†). The elemental analysis results (Table S1†) further reveal that the composite is composed of C (74.65%), H (1.67%), N (6.30%), and O (calculated, 17.38%) atoms, which is consistent with the EDX results. All these results showed that the GA–NC-1 is high content of N doped graphitization carbon composites. Raman spectroscopy was used to confirm the quality of the GA–NC-1 (Fig. S5†). Two major features, a D band and G band, were observed at around 1385 and 1575 cm−1 respectively. The relative intensity of the “disorder” D-band and the crystalline G-band (ID/IG) for the GA–NC-1 is about 1.1, indicating its disorder and amorphous feature. The FTIR spectrum of GA–NC-1 was shown in Fig. S6.† Corresponds to the previous XPS result, the peak at 1121 cm−1 is caused by the vibration of C–O, covers the characteristic peak of graphite. The shoulder peak around 1542 cm−1 reflects the skeletal vibration of graphene and the aromatic clusters of NC. The peaks around 2925 cm−1, which corresponds to the C–H stretching mode, suggest that the NC may be broken into small pieces with more edges which also correspond to the amorphous nature of GA–NC-1. Therefore, we can conclude that the as-prepared GA–NC-1 is made up of the graphene sheets coated with N-doped amorphous carbon which is mainly composed of pyridinic N and graphitic CN species derived from the dehydration and carbonization of chitosan. Thus, this strategy provides a feasible way to build up 3D porous carbons decorated graphene composites with a high level of N doping.
 | ||
| Fig. 2 XPS spectra of the GA–NC-1. (a) Survey spectrum. (b) C1S spectrum. (c) N1S spectrum. (d) O1S spectrum. | ||
Given the unique porous features and the heteroatom doped carbons of the composites, their electrochemical performance as Electrochemical Capacitive Energy Storage were evaluated in a three-electrode system, applying 6 M KOH as electrolyte. Fig. 3a displays the galvanostatic charge–discharge curves for the electrodes a charge–discharge current density of 0.5 A g−1. All of the obtained galvanostatic charge–discharge curves were nonsymmetric triangular shape, which is the signal of the existence of pseudocapacitance besides electric double layer capacitance. It should be attributed to the N-doped carbon and the remaining oxygen-containing groups in the composites. The specific capacitances of the composites electrodes in terms of scan rate were shown in Fig. 3b. It can be clearly seen that the composites have higher specific capacitance than NC at all scan rates which is much better than the pristine GA, suggesting the intimate contact between the electrolyte and the active material. And this intimate contact results in excellent charge transfer kinetics due to the porous and cross-linked structure formed. The RGO in the composites serve as multidimensional pathways to facilitate the transport of electrons in the bulk electrode, and the NCs which provide enough micropores can facilitate transport of the electrolyte ions within the electrode materials. Nyquist plots of the supercapacitors were obtained by a frequency response analysis (FRA) of the frequency range from 10 mHz to 100 kHz (Fig. 3c). All the devices have the similar intercept on the real axis at high frequency (Fig. 3c, inset), which means that the intrinsic internal resistance of the electrode material and electrolyte is not the major determinant for the overall internal resistance of devices.26 But, compared with the pristine NC, the Nyquist plots of the composites based supercapacitors exhibit much smaller diameter of semicircular at the high to midfrequency region, which indicates that the RGO can effectively improve the conductively of NC. In addition, the composites based supercapacitor has a relatively shorter 45° Warburg region at the middle frequency, and a more vertical line at low frequencies, which indicates that the device has much higher ionic conductivity and better electrochemical performance. Fig. 3d shows the cycling lifetime tests over 1000 cycles for the samples at a current density of 5 A g−1. All the samples exhibited good cycling stability and maintained 99% of its initial capacitance even after 1000 cycles. It was demonstrated that the repetitive charge–discharge cycles did not result in any obvious changes of the structure of the samples, and simultaneous exfoliation of the electrode material from the nickel foam current collector did not occur. Obviously, GA–NC-1, among all the composites, shows the best electrochemical performance as Electrochemical Capacitive Energy Storage.
 | ||
| Fig. 3 (a) Galvanostatic charge–discharge curves of NC (0.56 g cm−3), GA–NC-0.5 (0.73 g cm−3), GA–NC-1 (0.66 g cm−3), GA–NC-1.5 (0.65 g cm−3), GA (1.15 g cm−3) supercapacitor at a charge–discharge current density of 0.5 A g−1. (b) Specific capacitance for NC, GA–NC-0.5, GA–NC-1 and GA–NC-1.5 at different constant currents. The specific capacitance value was obtained from the galvanostatic charge–discharge measurement. (c) Nyquist impedance plots at a frequency range of 10 mHz to 100 kHz. The inset shows a magnified view of the high frequency region of the impedance spectra. (d) The cycling performance of the supercapacitors with NC, GA–NC-0.5, GA–NC-1 and GA–NC-1.5 at a charge–discharge current density of 5 A g−1 over 1000 cycles. | ||
Fig. 4 shows the detailed electrochemical performance of GA–NC-1. As shown in Fig. 4a, the GA–NC-1-based electrodes exhibited a relatively rectangular shape even at 50 mV, indicating that an efficient electrochemical double-layer capacitors is established with high charge–discharge rates and ideal capacitive behavior. Fig. 4b displays the galvanostatic charge–discharge curves for the GA–NC electrodes at different current density. All of the obtained galvanostatic charge–discharge curves were nonsymmetric triangular shape, which is also the signal of the existence of pseudocapacitance besides electric double layer capacitance as described before. On the basis of the discharging curve line, the specific gravimetric capacitance of GA–NC-1 was calculated to be 226 F g−1 at 0.5 A g−1 which is the best of the samples (the detailed electrochemical performance of the other samples was shown in Fig. S7†). Obviously, the GA–NC shows better electrochemical performance than most of the activated carbons which have the capacitance of ∼100 F g−1 in aqueous electrolytes34 (Table S2†).
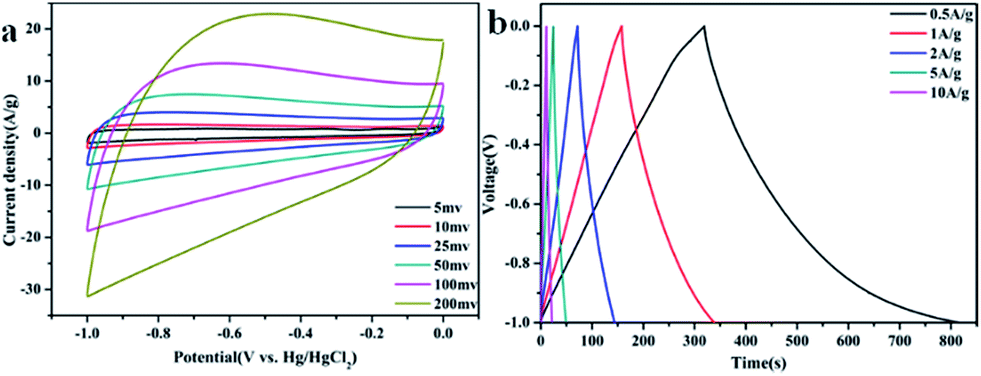 | ||
| Fig. 4 (a) Cyclic voltammetry curves obtained at different scan rates for GA–NC-1. (b) Galvanostatic charge–discharge curves of GA–NC-1 supercapacitor under different constant currents. | ||
One of the most important applications of heteroatom doped porous carbons is used as metal-free catalysts for the oxygen reduction reaction (ORR).34–36 Fig. 5 shows the ORR cyclic voltammograms curves (CVs) of the samples in O2 and N2-saturated 0.1 M KOH solution. The peak potential of the samples is around −0.3 V, suggesting these samples can be an effective electrocatalyst for ORR. Among them, GA–NC-1 showed an ORR onset potential of −0.18 V and a peak potential of −0.28 V versus Ag/AgCl, values the lowest of all the samples. For the supercapacitor and ORR applications, the RGO aerogel in GA–NC-1 is expected to not only act as a long distance 3D charge transporter, but also be a miniature current collector during the electrochemical processes, by taking advantage of its high electrical conductivity and 3D electron transport feature.
 | ||
| Fig. 5 ORR cyclic voltammograms curves (CVs) of the samples in O2 and N2-saturated 0.1 M KOH solution. | ||
Conclusions
In summary, we have proposed a facile graphene inspired strategy for the synthesis of microporous N-doped carbon coated 3D graphene based frameworks. Large amount of chitosan added into the system act as coatings to adhere on the graphene sheets and separate the graphene. The resulting GA–NC possesses macro- and microporous structures with a high level of N doping (6.30%). A π–π stacking interactions inspired forming mechanism was proposed to explain the formation process and the restricted thickness of the NC wall. The fabricated GA–NC show enhanced supercapacitive performance comparing with most of activated carbons and high efficiency as metal-free catalysts for ORR. It is expected that our synthetic strategy will offer the opportunity to build up various 3D hierarchical porous carbon materials for a variety of application, such as ECs, batteries, electronics, and sensors.Acknowledgements
This work was financially supported by National Science Foundation of China (NSFC) (no. 20934002, 51073043) and the National Basic Research Program of China (no. 2009CB930000).Notes and references
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed
.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS PubMed
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed
- H. Hu, Z. Zhao, W. Wan, Y. Gogotsi and J. Qiu, Adv. Mater., 2013, 25, 2219–2223 CrossRef CAS PubMed
- L. Xiao, D. Wu, S. Han, Y. Huang, S. Li, M. He, F. Zhang and X. Feng, ACS Appl. Mater. Interfaces, 2013, 5, 3764–3769 CAS
- Z. Chen, C. Xu, C. Ma, W. Ren and H.-M. Cheng, Adv. Mater., 2013, 25, 1296–1300 CrossRef CAS PubMed
- D. A. C. Brownson, L. C. S. Figueiredo-Filho, X. Ji, M. Gomez-Mingot, J. Iniesta, O. Fatibello-Filho, D. K. Kampouris and C. E. Banks, J. Phys. Chem. A, 2013, 1, 5962–5972 CAS
- H.-B. Yao, J. Ge, C.-F. Wang, X. Wang, W. Hu, Z.-J. Zheng, Y. Ni and S.-H. Yu, Adv. Mater., 2013, 25, 6691 CrossRef
- H.-B. Yao, J. Ge, C.-F. Wang, X. Wang, W. Hu, Z.-J. Zheng, Y. Ni and S.-H. Yu, Adv. Mater., 2013, 25, 6692–6698 CrossRef CAS PubMed
- Y. Xu, Q. Wu, Y. Sun, H. Bai and G. Shi, ACS Nano, 2010, 4, 7358–7362 CrossRef CAS PubMed
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS PubMed
- C. Hu, X. Zhai, L. Liu, Y. Zhao, L. Jiang and L. Qu, Sci. Rep., 2013, 3, 2065 Search PubMed
- G. Zhou, L.-C. Yin, D.-W. Wang, L. Li, S. Pei, I. R. Gentle, F. Li and H.-M. Cheng, ACS Nano, 2013, 7, 5367–5375 CrossRef CAS PubMed
- Z.-S. Wu, Y. Sun, Y.-Z. Tan, S. Yang, X. Feng and K. Muellen, J. Am. Chem. Soc., 2012, 134, 19532–19535 CrossRef CAS PubMed
- S. Yang, X. Feng, L. Wang, K. Tang, J. Maier and K. Muellen, Angew. Chem., Int. Ed., 2010, 49, 4795–4799 CrossRef CAS PubMed
- Q. Qu, S. Yang and X. Feng, Adv. Mater., 2011, 23, 5574–5580 CrossRef CAS PubMed
- Y. Liang, Y. Li, H. Wang and H. Dai, J. Am. Chem. Soc., 2013, 135, 2013–2036 CrossRef CAS PubMed
- Y. Su, S. Li, D. Wu, F. Zhang, H. Liang, P. Gao, C. Cheng and X. Feng, ACS Nano, 2012, 6, 8349–8356 CrossRef CAS PubMed
- Y. Feng, T. He and N. Alonso-Vante, Chem. Mater., 2007, 20, 26–28 CrossRef
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed
- T. F. Jaramillo, J. Bonde, J. Zhang, B.-L. Ooi, K. Andersson, J. Ulstrup and I. Chorkendorff, J. Phys. Chem. C, 2008, 112, 17492–17498 CAS
- X. Zhuang, F. Zhang, D. Wu, N. Forler, H. Liang, M. Wagner, D. Gehrig, M. R. Hansen, F. Laquai and X. Feng, Angew. Chem., 2013, 125, 9850–9854 CrossRef
- M. Li, J. Ding and J. Xue, J. Mater. Chem. A, 2013, 1, 7469–7476 CAS
- S. Yang, X. Feng, X. Wang and K. Müllen, Angew. Chem., Int. Ed., 2011, 50, 5339–5343 CrossRef CAS PubMed
- D. Krishnan, K. Raidongia, J. Shao and J. Huang, ACS Nano, 2013, 8, 449–457 CrossRef PubMed
- C.-C. Ji, M.-W. Xu, S.-J. Bao, C.-J. Cai, Z.-J. Lu, H. Chai, F. Yang and H. Wei, J. Colloid Interface Sci., 2013, 407, 416–424 CrossRef CAS PubMed
- L. Zhang, F. Zhang, X. Yang, G. Long, Y. Wu, T. Zhang, K. Leng, Y. Huang, Y. Ma, A. Yu and Y. Chen, Sci. Rep., 2013, 3, 1408 Search PubMed
- M. Sevilla and A. B. Fuertes, Chem.–Eur. J., 2009, 15, 4195–4203 CrossRef CAS PubMed
- M.-M. Titirici and M. Antonietti, Chem. Soc. Rev., 2010, 39, 103–116 RSC
- N. Baccile, G. Laurent, F. Babonneau, F. Fayon, M.-M. Titirici and M. Antonietti, J. Phys. Chem. C, 2009, 113, 9644–9654 CAS
- L. Zhao, N. Baccile, S. Gross, Y. Zhang, W. Wei, Y. Sun, M. Antonietti and M.-M. Titirici, Carbon, 2010, 48, 3778–3787 CrossRef CAS PubMed
- M.-M. Titirici, R. J. White, C. Falco and M. Sevilla, Energy Environ. Sci., 2012, 5, 6796–6822 Search PubMed
- E. Frackowiak and F. Béguin, Carbon, 2001, 39, 937–950 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Photographs of the samples, SEM and TEM images of the samples, isotherm plot of the samples, element analysis of chitosan and GA–NC, cyclic voltammetry curves and galvanostatic charge–discharge curves of the samples. See DOI: 10.1039/c4ra08945f |
| This journal is © The Royal Society of Chemistry 2014 |