
Photocatalytic reduction of CO2 to CO utilizing a stable and efficient hetero–homogeneous hybrid system†
Abstract
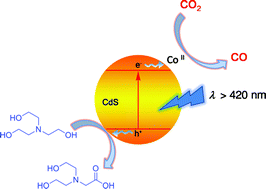
The combination of the conventional inorganic semiconductor CdS and a homogeneous catalyst could reduce CO2 to CO economically under visible light with high selectivity, stability, and efficiency.
 Please wait while we load your content...
Please wait while we load your content...

