
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular interactions of DNA with transfectants: a study based on infrared spectroscopy and quantum chemistry as aids to fluorescence spectroscopy and dynamic light scattering analyses†
A.
Lucotti
ab,
M.
Tommasini
*ab,
D.
Pezzoli
b and
G.
Candiani
*abc
aDepartment of Chemistry, Materials and Chemical Engineering “G. Natta”, Politecnico di Milano, via Mancinelli 7, 20131 Milano, Italy. E-mail: matteo.tommasini@polimi.it; gabriele.candiani@polimi.it
bResearch Unit Milano Politecnico, National Interuniversity Consortium of Materials Science and Technology – INSTM, via Mancinelli 7, 20131 Milano, Italy
cCentro Interuniversitario di Ricerca in Biotecnologie Proteiche “The Protein Factory”, Politecnico di Milano, CNR-ICRM Milano and Università degli Studi dell'Insubria, Milano, Italy
First published on 30th September 2014
Abstract
Cationic polymers are promising non-viral agents because of their ease of use, inexpensiveness and favorable safety profile. Being inherently cationic, they do spontaneously assemble in water with nucleic acids to give polyplexes. The more popular techniques and methods routinely applied for the characterization of such nanosupensions, unfortunately, do not afford direct observation of anion–cation interactions. We thus tackle the issue of probing at the molecular level the interplay occurring between the PO4− groups of double stranded DNA and the NH3+ groups of the simple and representative transfectant poly(L-lysine). By means of IR spectroscopy and Density Functional Theory (DFT) calculations we assigned the red shift of the anti-symmetric PO stretching wavenumber to the intermolecular water-mediated NH3+–PO4− interaction. These changes observed in moving from ineffective, low nitrogen (N) to DNA phosphate (P) ratios (N/Ps) to more effective conditions are in overall good agreement with changes in polyplex behavior unveiled by means of other analytical techniques such as dynamic light scattering, laser Doppler micro-electrophoresis, and fluorescence spectroscopy, but provide direct insight into the basic intermolecular DNA:transfectant interplay. The application of IR spectroscopic analysis to other non-viral gene delivery vectors may become an appealing approach to disclose molecular details on PO4−–cationic polymer interaction, taking into account conformational constraints and steric hindrance effects that the other techniques actually overlook.
Introduction
The use of nucleic acids as drugs for the treatment of a plethora of genetic and acquired diseases that conventional medicine fails to cure is collectively known as gene therapy. Since nucleic acids by themselves cannot be efficiently internalized by cells, one of the major challenges that still stands in the way of the complete success of gene therapy is the development of effective and non-toxic delivery agents, namely viral and non-viral vectors.1Although genetically engineered viral vectors are at present the most efficient delivery systems, serious safety concerns have hampered their widespread use and prompted the scientific community to develop non-viral gene vectors.2 Since their first description as gene vectors in 1987 by Wu and Wu,3 cationic polymers are one of the most used classes of non-viral agents because of their easiness of use, inexpensiveness and favorable safety profile. As these gene delivery vectors are inherently cationic, they do spontaneously interact with polyanionic nucleic acids to form nano-to-micro-scaled complexes called polyplexes. Polymer/DNA complexes, in turn, have to be taken up by cells and unpacked intracellularly to release their nucleic acid cargo that undergoes transcription and ultimately translation (i.e. transfection).4 Unfortunately, the in vivo application of polymeric gene vectors is rather limited because of their still low transfection efficiency caused by one or more bottlenecks among those mentioned here above. In order to improve their effectiveness, a number of studies dealing with establishing structure-function relationships have been reported.5–8 Detailed and extensive characterization of the physicochemical and biophysical features of polyplexes does basically rely on multiple techniques and methods, in first place dynamic light scattering (DLS) and electron microscopy (EM) for size measurements,9–11 Doppler velocimetry for surface charge measurements, while agarose gel retardation analysis and dye-exclusion assays for the determination of the binding strength between the polymers and the nucleic acids and the stability of polyplexes over time and in different environments.12–14 Unfortunately, mechanistic insights into polyplex formation and disassembly have been hampered by a lack of atomic resolution, and structural information on polymer/DNA complexes.10 On these premises, finding an effective gene delivery solution remains mostly a trial and error process.15
It is known that vibrational states depend on molecular structure and intermolecular interactions; hence, within this framework, IR spectroscopy might provide useful molecular information on DNA complexation that other techniques overlook. Furthermore, the assignment and interpretation of changes in vibrational spectra is nowadays fostered by the use of reliable quantum chemical tools, among which Density Functional Theory (DFT) framework provides the most effective balance between accuracy and computational cost.16 This allows considering sizable molecular models to tackle the theoretical interpretation of vibrational spectra of biologically relevant molecules, as we describe in this work. Instead of the overall view of polyplexes that many other techniques for characterizing colloidal dispersions allow for, the joint use of DFT and IR spectroscopy provides a molecular picture of the interactions occurring within polyplexes, which is experimentally probed by the spectral changes of a selected IR transition (anti-symmetric PO stretching) caused by the interaction of PO4− groups of the DNA with the NH3+ groups of poly(L-lysine) (PLL). Interestingly we noticed that this interaction is modulated by the presence of bridging water molecules. PLL was chosen as model transfectant because it is the oldest and a structurally simple (i.e. with only primary amines, the ε-amino groups) cationic polymer for gene delivery and it has been largely investigated in the literature over the last almost three decades.3,17
In this study, we compared different dye-based and label-free techniques to evaluate the DNA-complexation behavior of PLL. Our results lead to the proposal of using IR spectroscopy for the investigation of DNA complexation by cationic polymers, possibly paving the way toward the application of this technique for the evaluation of polyplex stability. Furthermore, DFT calculations were shown very successful in describing the spectroscopic effects of the interaction between the DNA and cationic polymers within polyplexes.
Results and discussion
In vitro transfection and cytotoxicity of PLL-based polyplexes
It is a matter of fact that the performance of a given gene delivery vector is a function of the nitrogen (N) to plasmid DNA phosphate (P) ratio (N/P) of use.1,7,13 Thus, we first evaluated the transfection activity of PLL as a function of N/P. Transfection experiments were carried out on HeLa and SKOV-3 cell lines in culture medium supplemented with 10% FBS, commonly used in in vitro experiments to take into account the effect of serum on gene vectors prior to animal studies.18 In fact, it is now renown that non-viral gene vectors in biological fluids such as plasma or serum, interact with proteins forming a dynamic protein shell called protein corona that can impair polyplex stability and often adversely impacts their behaviour.19,20 As expected, transfection efficiency increased progressively with raising the N/P (Fig. 1). At N/P 0.5 the luciferase signal was below the threshold of sensitivity of the assay, while significant transgene expression was observed only at N/P ≥ 1.5. The maximum transfection levels were obtained at N/P 3 (p < 0.05 vs. all lower N/Ps), beyond which a further increase in N/P resulted in a mere increase of the cytotoxicity levels (Fig. 1). Noteworthy, in all the transfection conditions tested, the cytotoxicity was very low in both the cell lines (<10%), thus not impairing transfection results.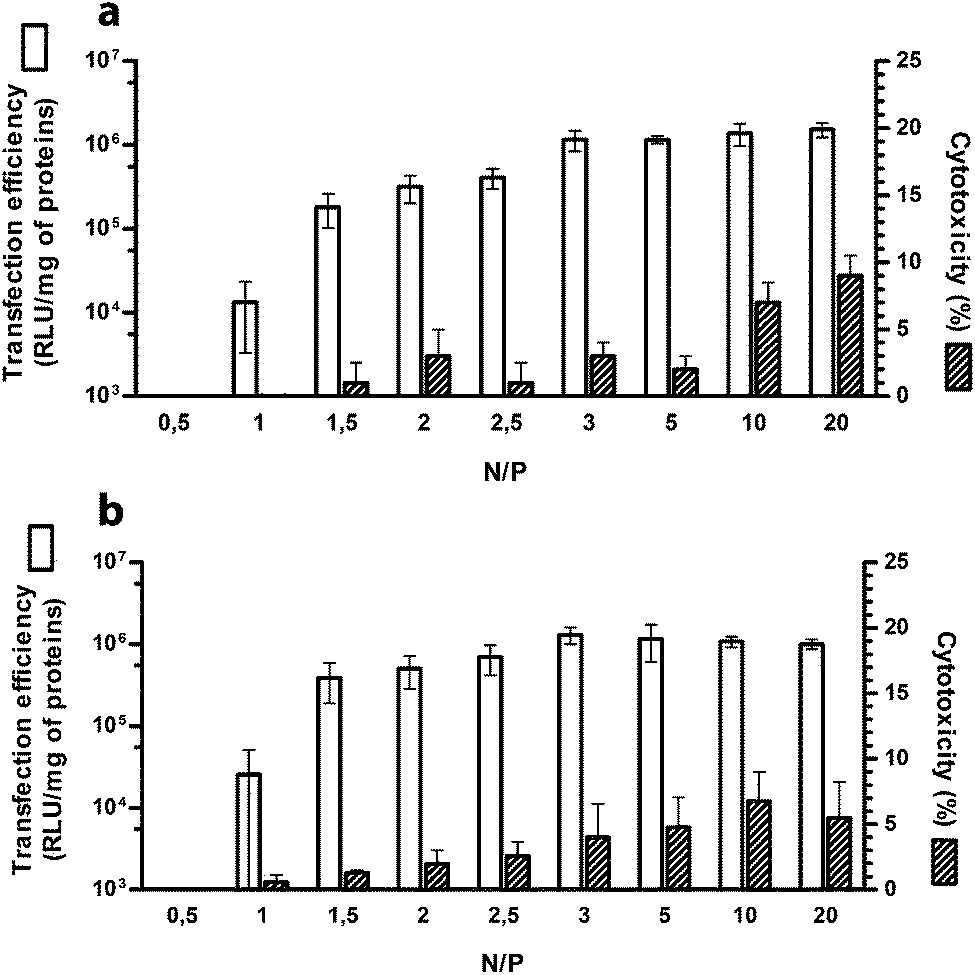 | ||
| Fig. 1 Transfection efficiency (white bars) and cytotoxicity (striped bars) of PLL-based polyplexes over a wide range of nitrogen (N) to plasmid DNA phosphate (P) ratios (N/Ps) and assayed on (a) HeLa and (b) SKOV-3 cell lines. Results are expressed as mean ± standard error of the mean (n = 4). | ||
Size and ζ-potential of PLL-based polyplexes
For efficient gene delivery it is fundamental that the polymers bind to and condense the extended structure of the DNA to form relatively tight complexes a few hundred nanometers in size and slightly positively charged on the surface. This is because after interacting with negatively charged cell membrane components, particles smaller than 1 μm in diameter can be internalized by non-phagocytic cells via endocytosis.1We thus evaluated the size (average hydrodynamic diameter, DH) and the overall surface charge (ζ-potential, ζP) of PLL-based polyplexes by DLS and laser Doppler micro-electrophoresis, respectively, as a function of the N/P. These techniques unfortunately do not provide any information about PLL–DNA interactions at the very molecular level.
As shown in Fig. 2, ζP curves as a function of N/P displayed a typical sigmoidal shape with the charge-inversion point (i.e. 0 mV) at N/P ≈ 1.25 at which a spike in the DH curve occurred.21 This neutralization or charge reversal phenomenon (also known as “overcharging” effect) results in an isoelectric point (IEP) and rapid aggregation of the colloidal uncharged polyplexes.
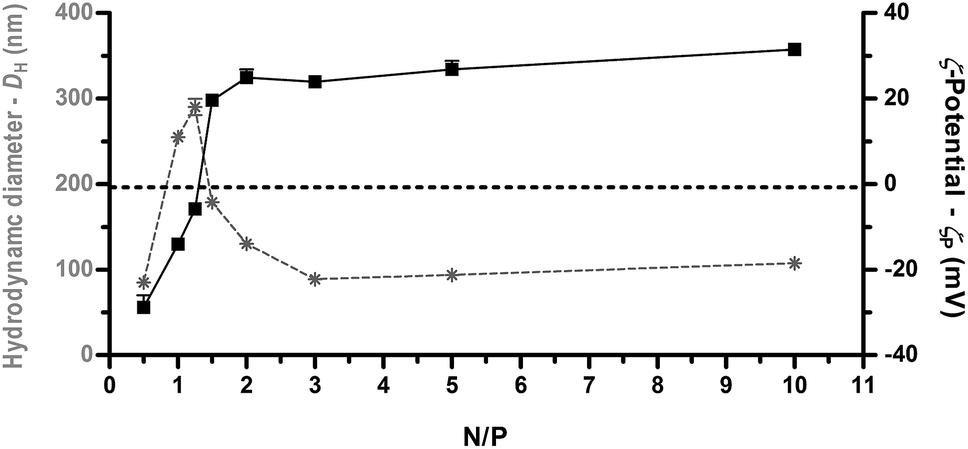 | ||
| Fig. 2 Average hydrodynamic diameter (DH, grey asterisks and dotted lines) and ζ-potential (ζP, full black squares and lines) of PLL-based polyplexes determined over a wide range of nitrogen (N) to plasmid DNA phosphate (P) ratios (N/Ps). Results are expressed as mean ± standard deviation (n = 4). | ||
The slight displacement of charge-inversion point from the theoretical N/P of 1 to the experimental 1 < N/P < 1.5, suggests that more than a NH3+ group on the polyelectrolyte chain is necessary to neutralize a single PO4− on the double-stranded DNA (dsDNA), and this can be ascribed to steric hindrance and conformational constraints of the PLL chain. Beyond the IEP, further addition of polycations leads to continuous polymer adsorption onto the polyplex surface, charge reversal, and restabilization of the particle suspension.22 Accordingly, we found that beyond N/P ≥ 2, a single unimodal population of polyplexes (polydispersity index ≈ 0.2) was observed with a DH of ≈ 100 nm and ζP > +20 mV. Inversely, a reliable estimation of the diameter of uncomplexed plasmid DNA (pDNA) could not be determined by DLS, as previously indicated also by others.23
Complexation ability of PLL
Fluorescence spectroscopy is a powerful technique with utility for studying the dynamic interactions and visualization of macromolecular complexes.24 Due to the excellent sensitivity of fluorescence compared to other optical techniques and to the large variety of fluorescent intercalating dyes available in the market, spectrofluorimetric steady-state analyses of DNA-dyes are particularly suited to investigate the DNA-condensation ability of gene delivery vectors.7,25 We evaluated by fluorophore-exclusion bio-analytical assay using SYBR Green I as DNA dye the ability of PLL to complex and condense nucleic acids as a function of N/P. Although very useful in providing fundamental information about the affinity between a polycation and the DNA, this technique does not allow direct identification of the interacting atoms and/or functionalities.A dramatic increase in brightness (>1000-fold) is observed when the unsymmetrical cyanine dye SYBR Green I does intercalate the dsDNA helix.24 When dsDNA is condensed by a polymer, the intercalating dyes are hindered in their access to the intercalation sites. Thus, the extent of formation of polyplexes can be inferred from the relative fluorescence of unassociated DNA after exposure of the polymer/DNA complexes to SYBR Green I.
As shown in Fig. 3, the amount of free DNA decreased as the N/P increased. Specifically, we observed that PLL can effectively complex the DNA already at very low N/P, with residual fluorescence levels lower than 10% with respect to naked DNA already for N/P ≥ 1, in roughly good agreement with what we obtained by DLS analysis and laser Doppler micro-electrophoresis. It is worth noting that the maximum quenching of the fluorescence signal (Fig. 3) was achieved immediately beyond the charge-neutralization point (i.e. at ζP ≈ 0 mV for N/P 1.25 and higher, Fig. 2) when the NH3+ charges of the PLL did counterbalance and neutralize the PO4− on the dsDNA.
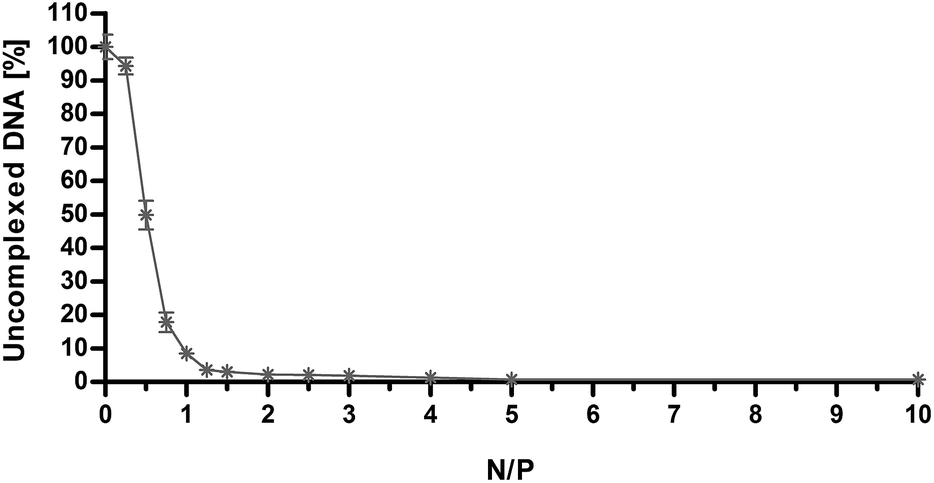 | ||
| Fig. 3 Fluorophore-exclusion titration of PLL as a function of nitrogen (N) to plasmid DNA phosphate (P) ratio (N/P). DNA complexation ability of PLL was evaluated by monitoring the exclusion of SYBR Green I from polyplexes. Results are expressed as mean ± standard deviation (n = 3). | ||
Analysis of phosphate stretching signals in the IR absorption of DNA
To get a direct evidence of the existence of interactions between the DNA phosphates and PLL amines and to gain more knowledge on how these interactions occur in such polyplexes we carried out a series of IR measurements on PLL interacting with DNA at N/Ps in the range of 0 to 5. The IR spectra of these different samples are displayed in Fig. 4. The assignment of symmetric (≈1090 cm−1) and anti-symmetric (≈1220 cm−1) PO stretching IR bands of the DNA is well established.26–28 Upon effective complexation, which, depending on the investigation technique considered was found to happen for N/P ≥ 1, we observed a red shift of the anti-symmetric PO stretching of about 4 cm−1. Once complexation has occurred no further PO peak red shift was observed. Based on the discussion above, we may deduce that the local environment nearby the PO4− of DNA has reached a stable structure with respect to the interacting species which is not affected any further by an increase of the N/P. Consequently, we can infer that IR measurements on PLL/DNA can proficiently recognise DNA complexation states adequate for transfection (i.e. N/P ≥ 1) but cannot identify polyplexes with the highest efficiency (i.e. N/P ≥ 3). On the other hand, IR is a probe-free technique that yields atomistic information not obtainable by fluorescent dye measurements. | ||
| Fig. 4 IR spectra of a sample of salmon sperm DNA (0.7 mg mL−1) in water solution of PLL at different nitrogen (N) to plasmid DNA phosphate (P) ratios (N/Ps) (reported for each spectrum). The red-shift of about 4 cm−1 upon complexation is observed for N/P > 1. | ||
We have also carried out DFT calculations on selected model molecules to investigate in some details the vibrational dynamics of the PO4− groups and correlate the observed wavenumber shifts to interactions with specific molecular entities. This is relevant for the investigation of the basic interactions that exist in polyplexes between the inner molecular layer of cationic polymers facing the nucleic acids.
As starting model we have considered a section of the double helix of DNA comprising four bases, whose structure optimized with DFT is given below (Fig. 5). This model has a remarkable size for current quantum chemical methods and common computational facilities (125 atoms, 1697 basis functions). The expensive part of the calculation is the evaluation of the second derivatives of the energy with respect to nuclear displacements, which is necessary for obtaining the vibrational normal modes and the simulation of the IR spectrum. Hence, this DNA model was not practical for investigating the spectroscopic changes induced by PO4−–PLL interactions, but it could be profitably used to define smaller sub-models focused on the PO4− group.
 | ||
| Fig. 5 The fully optimized equilibrium structure of the model DNA section considered for investigating the phosphate stretching vibrations. The model represents two short sections of interacting DNA strands. One strand is a cytosine-adenine (CA) sequence and the other is the complementary sequence (guanine-thymine – GT). F denotes furanose rings.‡ | ||
In Fig. 6 we present the comparison between the experimental IR spectrum of the DNA in water and the simulated spectrum obtained from DFT calculations carried out on the model of Fig. 5. The overall agreement between simulation and experimental observation is remarkably good, after taking into due consideration the fact that the DNA model is a significant simplification of the real sample. In particular, all main features in the experimentally accessible range (1400–950 cm−1) find a theoretical counterpart, after accepting an inevitable wavenumber shift and relative intensity adjustment.
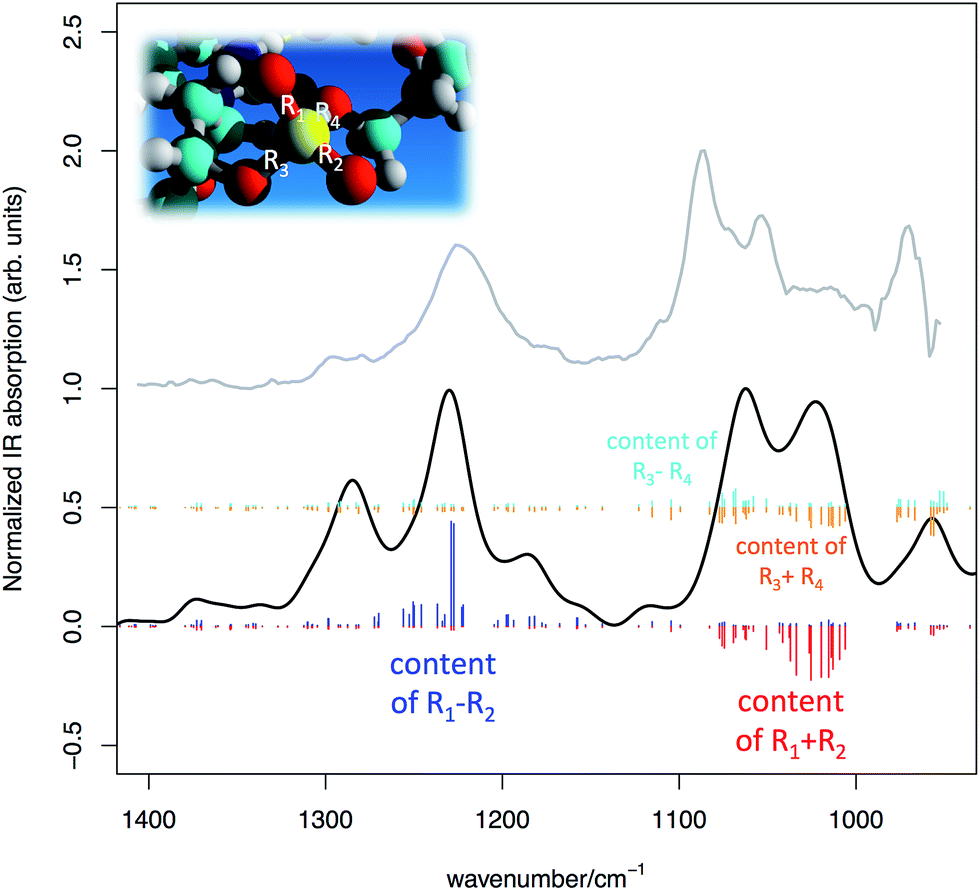 | ||
| Fig. 6 Simulated IR spectrum of the DNA model represented in Fig. 5 (DFT calculations, simulated FWHM = 30 cm−1) (black line) compared with the experimental IR absorption of a sample of salmon sperm DNA in water solution (grey line). Sticks of different colours represent collective PO stretching contributions from the phosphate groups (see text for details). | ||
The model also provides a detailed description of the normal modes, which foster the understanding of the spectroscopic features associated to the PO4− groups. In particular two aims are reached:
(i) the assignment of the PO symmetric/anti-symmetric stretching bands shows that the PO stretching content is actually shared among several normal modes in the expected spectral regions. This is the effect of vibrational coupling existing between PO4− and the nearby groups (see Fig. 6);
(ii) based on the inspection of the coupling existing between phosphate and nearby groups (i) one can use the structure of the DNA backbone (taken from Fig. 5) to define a smaller sub-model aimed at describing the effects of interaction of the PO4− with cationic species of interest in transfection.
Following point (ii) we have considered a section of DNA backbone comprising two furanose rings connected by one PO4− group. This model is shown in Fig. 7 and labeled (a). To model the interaction effect of the cationic transfectant considered in our experiments (PLL) we have considered just the NH3+ and a section of the connected alkyl chain (C4H8), namely CH3–CH2–NH3+, and placed it in close vicinity of the PO4− group of the backbone (model (b) in Fig. 7). The effect of anion–cation interaction on the anti-symmetric PO stretching is dramatic, as the comparison provided in Fig. 7 clearly shows. Upon interaction a marked red shift of about 40 cm−1 is observed.
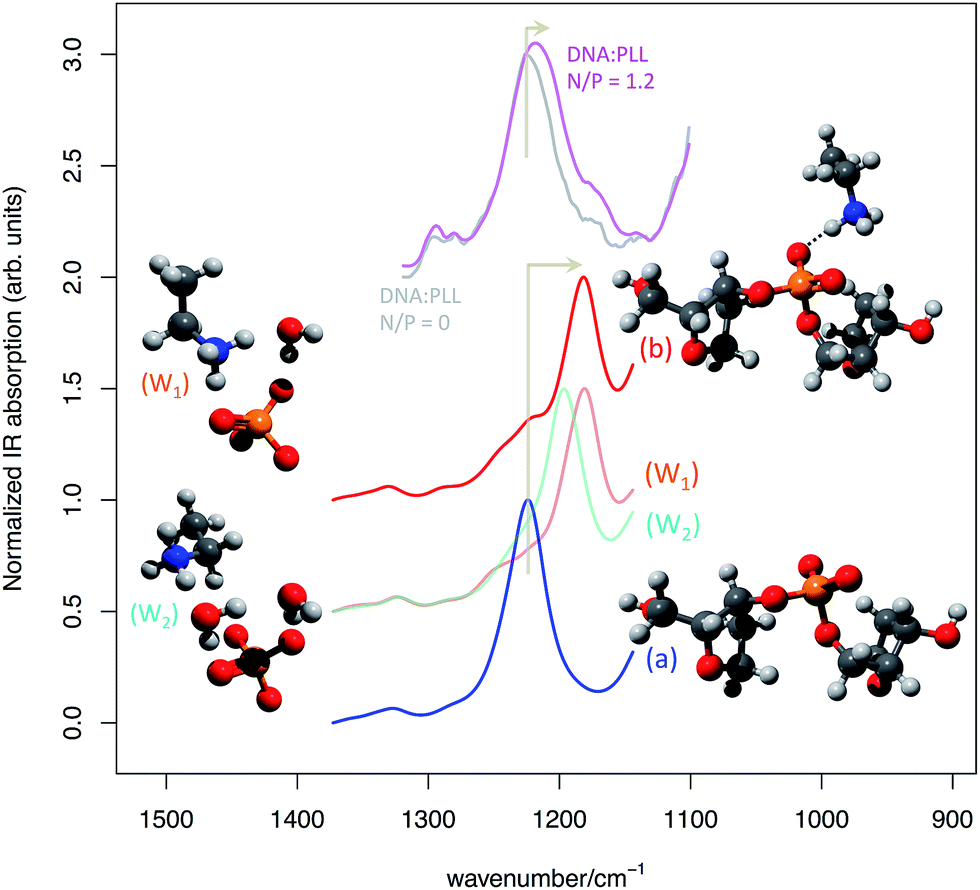 | ||
| Fig. 7 Simulated IR spectrum of (a) the non-interacting and (b) interacting DNA backbone model extracted from the molecular model represented in Fig. 4 (from DFT calculations, simulated FWHM = 30 cm−1). To ease comparison with experiment a scaling factor of 1.04 has been applied to the computed wavenumber values. The experimental IR absorption spectra of a sample of salmon sperm DNA in water solution of PLL at N/P 0 (grey line) and N/P 1.2 (magenta line) are also reported for comparison. For simplicity of representation just a section of models W1 and W2 is represented, but the models considered in the calculations are similar to (a) and (b) on the backbone side. | ||
From the experimental point of view, the red shift of the anti-symmetric phosphate stretching is more limited (see Fig. 7). This is explained by the fact that not all PO4− groups, even at a N/P 1.2, are actually able to strongly interact with the NH3+ groups as in model (b). Hence the experimental spectrum is the result of the average signal produced by interacting and non-interacting phosphates, which reduces the observed red shift with respect to the theoretical expectations of model (b). The water environment typical of transfection scenarios requires considering also the explicit role of water molecules at the anion–cation interaction site.
For this reason, we have modified model (b) including one or two explicit water molecules in mediating the anion–cation interaction (model (W1) and (W2), respectively).
The presence of water molecules screening the direct NH3+–PO4− interaction has a significant effect on the red shift of the anti-symmetric phosphate stretching, which is progressively reduced along the sequence (a) > (W1) > (W2) (see Fig. 7). Finally, we have considered a further model (W3) (not shown in Fig. 7, but reported in ESI†) that corresponds to model (W2) with the inclusion of one additional water molecule hydrogen-bonded to the phosphodiester oxygen of the PO4− group. Consistently with the trend discussed above, its associated anti-symmetric stretching lies between (W2) and (a) (Fig. SI1†).
Conclusions
By the joint use of several experimental and theoretical methods, this proof-of-concept study demonstrates that IR spectroscopy can be profitably used to investigate the DNA complexation by cationic polymers, allowing identifying transfection-active DNA complexation states by a red shift in the anti-symmetric (≈1220 cm−1) PO stretching IR band. Noteworthy, IR spectra allow definite discrimination between ineffective DNA binding (see N/P < 1 in Fig. 4) and transfection-active polyplexes (see N/P ≥ 1 in Fig. 4) as a function of the polycation concentration, as demonstrated by comparison with DNA complexation assay, size and ζP measurements. Hence we can conclude that IR spectroscopy can be used to probe DNA–cation interaction for applications in gene delivery development and in the large segment of the research involving processes where DNA interactions with biologically active molecules are very relevant events, such as in gene regulation studies. Furthermore we were able to predict by a simple DFT model the effect of anion–cation interaction on the IR spectrum, thus paving the way toward the use of this quantum mechanical tool for the investigation of the interactions occurring within polyplexes and for the prediction of their behaviour and stability in different environments.Based on the results presented here above, application of IR to other transfection scenarios becomes an appealing approach to disclose molecular details on the basic DNA–cation interaction. This is expected to depend on the selected cationic species and on the capability of the cationic polymer to effectively bind the DNA, which in turn depends on the molecular flexibility of the transfectant and its steric hindrance. Through the modulation of the anti-symmetric PO stretching wavenumber as a function of the N/P ratio one can monitor the combined effect of all these molecular properties, which are at the basis of transfection.
Experimental and methods
Materials
PLL hydrobromide (Mw: 4–15 kDa, as determined by viscosity) was purchased from Sigma-Aldrich (Milan, Italy). BCA Protein Assay Kit was from Thermo Fisher Scientific (Rockford, IL, USA). The Luciferase Assay System and the pDNA encoding for the modified firefly luciferase pGL3-Control Vector (5.2 kbp) were all from Promega (Milan, Italy). Salmon sperm DNA was purchased from Life Technologies Italia (Monza, Italy) at a concentration of 10 mg mL−1. All the other chemicals, solvents, and culture reagents used were purchased from Sigma-Aldrich if not differently specified.HeLa (human cervix carcinoma, CCL-2.2) and SKOV (human adenocarcinoma, HTB77) cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Fluorimetric, spectrophotometric and luminescence analysis were performed using GENios Plus reader (Tecan, Segrate, Italy).
Preparation of polyplexes
Polyplexes were prepared at room temperature (r.t.) by adding pGL3 or salmon sperm DNA to a solution of 4–15 kDa PLL in deionized water (dH2O) at the desired polymer concentration to yield different N/Ps, and incubated for 20 min at r.t. before use.SYBR Green I-exclusion assay
The ability of 4–15 kDa PLL to complex DNA was evaluated by fluorophore-exclusion titration assay, as previously described.13 Polyplexes were prepared in different volumes, depending on the N/P, using 400 ng of pGL3 and then diluted to 100 μL with 10 mM HEPES pH 7.0 containing 2× SYBR Green I (λex = 497 nm; λem = 520 nm). Fluorescence was measured using a microplate reader GENios Plus (Tecan, Segrate, Italy) and normalized over the fluorescence of naked pDNA.Measurement of size and surface charge of polyplexes
Size (DH) analyses and surface charge (ζP) measurements of polyplexes were performed by means of DLS and laser Doppler micro-electrophoresis using a Malvern Zetasizer Nano ZS instrument (Malvern, UK) fitted with a 633 nm laser at a fixed scattering angle of 173°. Polyplexes (100 μL, 2 μg of pGL3) were prepared as described above and were diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 in dH2O and left equilibrating at 25 °C prior to measurements.
:4 in dH2O and left equilibrating at 25 °C prior to measurements.
Cell culture and transfection
HeLa and SKOV-3 cells were cultured in humidified atmosphere of 5% CO2 at 37 °C. Cell culture medium was Dulbecco's Modified Eagle Medium (DMEM) containing 10 mM HEPES buffer, 1 mM sodium pyruvate, 100 U mL−1 penicillin, 0.1 mg mL−1 streptomycin, and 2 mM glutamine and supplemented with 10% (v/v) fetal bovine serum (FBS). The day before transfections, cells were plated in 96-well cell culture plates at a density of 2 × 104 cells per cm2. 102.4 ng per well of pGL3 were complexed as described above with 4–15 kDa PLL at the desired N/P and then added to cells in a final volume of 100 μL per well of medium. Twenty-four hours later cell viability was assessed using AlamarBlue cell viability assay (Life Technologies Italia, Monza, Italy) according to manufacturer's guidelines. Cytotoxicity was defined as 100% – viability [%]. Results were expressed as % relative to the untreated controls. Afterwards, cells were lysed with Cell Culture Lysis Reagent (Promega, Milan, Italy) and luciferase expression was quantified by Luciferase Assay System and normalized to the protein content in each sample, as determined by bicinchoninic acid (BCA) assay. Results were expressed as relative light units (RLU) per mg of proteins.Vibrational spectroscopy and quantum chemical calculations
IR spectra have been collected with Nicolet Nexus FT-IR interferometer. Water solutions of salmon sperm DNA and PLL have been measured using sealed cells with CaF2 windows and path length of 12 μm. Starting from the purchased salmon sperm DNA solution at 10 mg mL−1, we have prepared more diluted solutions at 0.7 mg mL−1 using deionized water. This choice has been dictated by the trade-off between two contrasting aims: the will of more closely approach the low concentration regime adopted in transfection, while keeping the IR signal of salmon sperm DNA strong enough to allow reliable measurements. Omnic software has been used for baseline subtraction and spectral averaging.Quantum chemical calculations have been carried out with the Gaussian09 quantum chemistry computer code29 on a small computer cluster operated at the Dept. of Chemistry, Materials and Chemical Engineering (Politecnico di Milano). We have considered a DFT method including dispersion forces, B97D/6-31+G(d,p), to account for π–π stacking interactions of the selected model DNA structure (Fig. 4). For models denoted in Fig. 6 as (a, b, W1, W2) we have included IEFPCM mean field correction for water solvation.30 This turns out to be essential for correctly modelling the interaction of the formal proton that is shared between the phosphate oxygen and the quaternary nitrogen.
By following the methodology described in the work by Radice et al.,31 we have analysed the Cartesian nuclear displacements associated to the normal modes of the selected molecular models in terms of valence internal coordinates, which directly relate the computed IR signals to vibrations of specific moieties in the chemical structure of the model molecule (such as, for instance, the stretching of a given PO bond). The output of DFT calculations typically reports normal modes information in terms of Cartesian nuclear displacements. This requires the application of a linear transformation (B) that converts Cartesian normal modes (Lx) into their internal coordinates counterpart (LR = BLx).31 In this way, for each given k-th normal mode, it is possible to determine the contribution to the displacement of the j-th valence internal coordinate as the matrix element (LR)jk. Further details on this procedure are given in ESI.†
In order to unequivocally assign symmetric PO stretching and anti-symmetric PO stretching we have considered the free PO bonds (R1, R2) of the two PO4− groups (A, B) of the DFT model reported in Fig. 4. This allows to define symmetric (+) and anti-symmetric (−) combinations, i.e. (R1 ± R2), locally to A and B. For a given k-th normal mode, the associated content of say, symmetric/anti-symmetric PO stretching on PO4− A, will be given by |(LR)ka ± (LR)kb| where a and b denote the indices of the PO stretching coordinates R1 and R2 of PO4− A (respectively). A completely similar approach is taken for the coordinates of PO4− B.
The final quantity, which is used in Fig. 5 to display the assignment of the computed IR features is given by the sum of the contributions from both PO4− groups, namely: |(LR)ka ± (LR)kb|A + |(LR)ka ± (LR)kb|B (where the + sign stands for the symmetric PO stretching and the – sign stands for the anti-symmetric PO stretching).
Furthermore we also consider the other two PO stretching coordinates of both PO4− (we name them R3, R4) that are directly connected to DNA backbone. Similarly to (R1, R2) we define in this case as well the symmetric and anti-symmetric combinations. It is possible to see that also the “backbone” PO bonds are participating to some extent to the symmetric PO stretching signal.
Geometry optimizations of models (a) and (b) have been carefully carried out to ensure that the structure of the backbone was kept the same as in the DNA model, since our aim is focused on the normal modes of the phosphate group. Hence just the atoms of the phosphate group and counter cation were left free to move during geometry optimizations. It is worth to mention that the models (a and b) do not allow to investigate with the same accuracy the other vibrational features of DNA other than the anti-symmetric PO stretching modes. For the former a greater involvement of furanose vibrations is expected and actually found. This is due to the fact that models (a) and (b), for the sake of simplicity, do not consider any DNA base connected to the sugar rings. Actually, in both (a) and (b) model we replace one nitrogen atom of the two DNA bases (CA) with a hydrogen atom. This is also fully relaxed together with the connected carbon during geometry optimization. Geometry optimization of models (W1), (W2), (W3) has been carried out similarly as models (a) and (b) with the additional optimization of the position of the explicit water molecules.
Statistical analysis
GraphPad version 5.04 (GraphPad software, La Jolla, CA, USA) was used for statistical analysis. Comparisons among groups were carried out by one-way analysis of variance (ANOVA). Significance was retained when p < 0.05. Data are expressed as mean ± standard error of the mean (SEM).Acknowledgements
We thank the Italian MIUR for financial support, under the auspices of the FIRB program (Futuro in Ricerca 2008, grant number RBFR08XH0H). We acknowledge Lisa Zuzolo for her involvement, during her Master Thesis, with the IR measurements described here. We acknowledge both referees for constructive discussion.Notes and references
- D. Pezzoli and G. Candiani, J. Nanopart. Res., 2013, 15, 1523 CrossRef.
- D. Pezzoli, R. Chiesa, L. De Nardo and G. Candiani, J. Appl. Biomater. Funct. Mater., 2012, 10, e82–91 Search PubMed.
- G. Y. Wu and C. H. Wu, J. Biol. Chem., 1987, 262, 4429–4432 CAS.
- H. Eliyahu, Y. Barenholz and A. J. Domb, Molecules, 2005, 10, 34–64 CrossRef CAS.
- W. T. Godbey, K. K. Wu and A. G. Mikos, J. Biomed. Mater. Res., Part A, 1999, 45, 268–275 CrossRef CAS.
- L. Wightman, R. Kircheis, V. Rossler, S. Carotta, R. Ruzicka, M. Kursa and E. Wagner, J. Gene Med., 2001, 3, 362–372 CrossRef CAS PubMed.
- D. Pezzoli, F. Olimpieri, C. Malloggi, S. Bertini, A. Volonterio and G. Candiani, PLoS One, 2012, 7, e34711 CAS.
- J. Hofman, M. Buncek, R. Haluza, L. Streinz, M. Ledvina and P. Cigler, Macromol. Biosci., 2013, 13, 167–176 CrossRef CAS PubMed.
- Y. Niebel, M. D. Buschmann, M. Lavertu and G. De Crescenzo, Biomacromolecules, 2014, 15, 940–947 CrossRef CAS PubMed.
- L. E. Prevette, E. N. Nikolova, H. M. Al-Hashimi and M. M. Banaszak Holl, Mol. Pharm., 2012, 9, 2743–2749 CrossRef CAS PubMed.
- A. L. Parker, D. Oupicky, P. R. Dash and L. W. Seymour, Anal. Biochem., 2002, 302, 75–80 CrossRef CAS PubMed.
- M. Wagner, A. C. Rinkenauer, A. Schallon and U. S. Schubert, RSC Adv., 2013, 3, 12774–12785 RSC.
- A. Ghilardi, D. Pezzoli, M. C. Bellucci, C. Malloggi, A. Negri, A. Sganappa, G. Tedeschi, G. Candiani and A. Volonterio, Bioconjugate Chem., 2013, 24, 1928–1936 CrossRef CAS PubMed.
- D. Fischer, T. Bieber, Y. X. Li, H. P. Elsasser and T. Kissel, Pharm. Res., 1999, 16, 1273–1279 CrossRef CAS.
- M. Brgles, M. Santak, B. Halassy, D. Forcic and J. Tomasic, Int. J. Nanomed., 2012, 7, 393–401 CrossRef CAS PubMed.
- V. Barone, A. Baiardi, M. Biczysko, J. Bloino, C. Cappelli and F. Lipparini, Phys. Chem. Chem. Phys., 2012, 14, 12404–12422 RSC.
- M. Mannisto, S. Vanderkerken, V. Toncheva, M. Elomaa, M. Ruponen, E. Schacht and A. Urtti, J. Controlled Release, 2002, 83, 169–182 CrossRef CAS.
- C. K. Goldman, L. Soroceanu, N. Smith, G. Y. Gillespie, W. Shaw, S. Burgess, G. Bilbao and D. T. Curiel, Nat. Biotechnol., 1997, 15, 462–466 CrossRef CAS PubMed.
- A. L. Barran-Berdon, D. Pozzi, G. Caracciolo, A. L. Capriotti, G. Caruso, C. Cavaliere, A. Riccioli, S. Palchetti and A. Lagana, Langmuir, 2013, 29, 6485–6494 CrossRef CAS PubMed.
- G. Caracciolo, F. Cardarelli, D. Pozzi, F. Salomone, G. Maccari, G. Bardi, A. L. Capriotti, C. Cavaliere, M. Papi and A. Lagana, ACS Appl. Mater. Interfaces, 2013, 5, 13171–13179 CAS.
- J. H. Jeong, S. H. Song, D. W. Lim, H. Lee and T. G. Park, J. Controlled Release, 2001, 73, 391–399 CrossRef CAS.
- J. Kleimann, C. Gehin-Delval, H. Auweter and M. Borkovec, Langmuir, 2005, 21, 3688–3698 CrossRef CAS PubMed.
- D. Y. Kwoh, C. C. Coffin, C. P. Lollo, J. Jovenal, M. G. Banaszczyk, P. Mullen, A. Phillips, A. Amini, J. Fabrycki, R. M. Bartholomew, S. W. Brostoff and D. J. Carlo, Biochim. Biophys. Acta, 1999, 1444, 171–190 CrossRef CAS.
- A. I. Dragan, R. Pavlovic, J. B. McGivney, J. R. Casas-Finet, E. S. Bishop, R. J. Strouse, M. A. Schenerman and C. D. Geddes, J. Fluoresc., 2012, 22, 1189–1199 CrossRef CAS PubMed.
- D. Pezzoli, M. Zanda, R. Chiesa and G. Candiani, J. Controlled Release, 2013, 165, 44–53 CrossRef CAS PubMed.
- M. Banyay, M. Sarkar and A. Graslund, Biophys. Chem., 2003, 104, 477–488 CrossRef CAS.
- Y. F. Guan and G. J. Thomas, Biopolymers, 1996, 39, 813–835 CrossRef CAS.
- Y. Guan and G. J. Thomas, Biophys. J., 1996, 71, 2802–2814 CrossRef CAS.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- S. Radice, M. Tommasini and C. Castiglioni, J. Mol. Struct., 2010, 974, 73–79 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra08845j |
| ‡ The starting structure used as input for subsequent DFT calculations as been obtained using tools available from: http://casegroup.rutgers.edu |
| This journal is © The Royal Society of Chemistry 2014 |