
Graphene–hemin hybrid nanosheets as a label-free colorimetric platform for DNA and small molecule assays†
Cui Hu,
Qiang Xi,
Jia Ge,
Feng-Yan Luo,
Li-Juan Tang*,
Jian-Hui Jiang and
Ru-Qin Yu*
State Key Laboratory of Chemo/Bio-Sensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha, 410082, P. R. China. E-mail: tanglijuan@hnu.edu.cn; rqyu@hnu.edu.cn; Fax: +86-731-88822577; Tel: +86-731-88822577
First published on 29th October 2014
Abstract
Here we report a novel label-free colorimetric biosensing strategy based on graphene–hemin hybrid nanosheets (GHs) for DNA and small molecule assays. GHs were synthesized by a simple wet-chemical method and they possess excellent properties from both graphene and hemin. When mixed with ssDNA, GHs exhibited enhanced catalytic activity in the presence of ABTS and H2O2. In contrast, the GHs mixed with dsDNA had lower catalytic activity than those mixed with ssDNA. On the basis of this property of the GHs, a novel universal label-free colorimetric method was proposed. This simple strategy has advantages over conventional assays because of its label-free design, extreme operational simplicity and low cost. DNA and cocaine were studied as model cases and the results revealed that the developed method had potential for becoming a universal platform for detection of a wide range of analytes.
Introduction
Since its recent appearance, a new hybrid nanosheet consisting of graphene oxide (GO) and hemin has become of interest to researchers in the field of biochemical sensing.1–5 As a one-atom-thick nanomaterial, GO has attracted a lot of attention for its excellent physical properties such as large surface area, good water-solubility and its ability to distinguish single-strand DNA (ssDNA) from double-strand DNA (dsDNA).6–8 Its fluorescence quenching ability for fluorophores is also superior to many other materials, which has made it one of the most commonly used nanomaterials for fluorescence biosensing in the last decade.9–11 However, the fluorescence methods are usually associated with disadvantages such as complicated labelling and high cost. Hemin (iron protoporphyrin), as the active center of heme-proteins, has peroxidase-like activity similar to the peroxidase enzyme, which could provide a colorimetric signal.12,13 Therefore, the graphene–hemin hybrid (GHs) possesses excellent properties from both graphene and hemin. Many studies focusing on the catalytic performance of GHs have been conducted.14–19 For example, hemin functionalized GO has served as a peroxidase probe for the sensitive detection of biomarkers,15–17 an effective oxygen reduction catalyst for oxidation of C–H bonds in organic compounds18 and as an agent for the sensitive quenching of fluorescent materials.19 Thus, this novel nanomaterial is expected to offer new avenues of investigation when it is applied to a particular application.Cocaine is one of the most dangerous illegally abused drugs to human health, due to its enormous impact on the central nervous system. For many years, sensitive and selective methods have been developed for the determination of cocaine.20–22 Since the aptamer of cocaine was discovered in 2000, a number of sensors based on the cocaine aptamer have been reported.23,24 Various amplification strategies have been used to improve the sensitivity, such as rolling circle amplification (RCA), polymerase chain reaction (PCR) and so on.25–28 However, in most cases, the amplification strategies have limitations owing to the need for sophisticated instrumentation, requirement of high technical expertise, as well as long analysis times. Therefore, it is of considerable interest to synthesize new materials possessing both peroxidase activity and DNA recognition ability in cocaine assays.
In this paper, GH nanosheets were synthesized through a simple wet-chemical method, they were initially found to be responsive to ssDNA and able to distinguish between ssDNA and dsDNA directly in the presence of 2,2′-azinobis-(3-ethylbenzthiazoline)-6-sulphonate (ABTS) and H2O2. When ssDNA was mixed with the GHs, the colorimetric signal intensity increased significantly. In contrast, the GHs mixed with dsDNA had a lower catalytic activity than their counterparts mixed with the ssDNA. Compared with a previous report, salt was not needed for the GHs to distinguish between ssDNA and dsDNA and the whole reaction process was homogeneous.1 Based on this finding, we proposed a novel label-free colorimetric strategy and constructed sensors for DNA and cocaine assays. Moreover, this proposed strategy had the potential to be practical due to its extreme simplicity and low cost.
Experimental section
Reagents
Graphene oxide was purchased from XF Nano Co. Ltd (Nanjing, China). Hemin, ABTS, TMB (3,3′,5,5′-tetramethylbenzidine) and hydrazine hydrate solution were obtained from Sigma-Aldrich (Missouri, USA). Cocaine, morphine hydrochloride (MHC), caffeine and theophylline were obtained from Beijing Institute for Drug Control (Beijing, China) and used without further purification. O-Phenylenediamine (OPD), H2O2 and NH3·H2O were supplied by Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). DNA oligonucleotides were obtained from Sangon Biotechnology Co. Ltd (Shanghai, China). The DNA sequences were as follows:P1 (probe):
ACCTGGGGGAGTATTGCGGAGGAAGGTTTTTT
T1 (complementary target):
AAAAAACCTTCCTCCGCAATACTCCCCCAGGT
T2 (single base mismatch target):
AAAAAACCTTCCTCCGCAATACTCCCCCAGCT
T3 (three base mismatch target):
ATAAAACCTTCCTCAGCAATACTCCCCCAGCT
T4 (unrelated DNA):
AGTCCGTGGTAGGGCAGGTTGGGGTGACTAGT
P2 (cocaine aptamer):
GGGAGACAAGGAAAATCCTTCAATGAAGTGGGTCTCCC
Deionized and sterilized water (resistance > 18.2 MΩ cm−1) was used throughout the experiments.
Apparatus
AFM images were taken through a multimode 8 atomic force microscope (Bruker, USA) and the surface was scanned at 1 Hz with a resolution of 512 lines per image. TEM images were obtained with a JEM-2100F high-resolution transmission electron microscope operating at 200 kV. Kinetic measurements were carried out by monitoring the absorbance at a corresponding wavelength on a Shimadzu UV-2450 UV-Vis spectrophotometer.Preparation of hemin functionalized graphene nanosheets (GHs)
GH nanosheets were synthesized according to the previously reported procedure.1 Hemin was assembled on the surface of graphene oxide sheets via π–π interactions between the porphyrin ring and the hexagonal cells of the graphene oxide. The homogenous graphene oxide (20.0 mL of 0.5 mg mL−1 dispersion) was mixed with 20.0 mL of 0.5 mg mL−1 hemin followed by drastic shaking for several minutes. Then 200.0 μL ammonia solution and 30.0 μL hydrazine hydrate were added to the solution. After being vigorously shaken for 1 h, the vial was put in a water bath (60 °C) for 24 h. The obtained black dispersion was then centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min to remove free hemin and the precipitated conjugates were redispersed in water followed by rinsing with water for three times. The acquired GHs could be redispersed in water to a final concentration of 10 μg mL−1. Moreover, the graphene oxide nanosheets were treated under identical conditions except no hemin was added, which obtained reduced graphene oxide (RGO).
000 rpm for 30 min to remove free hemin and the precipitated conjugates were redispersed in water followed by rinsing with water for three times. The acquired GHs could be redispersed in water to a final concentration of 10 μg mL−1. Moreover, the graphene oxide nanosheets were treated under identical conditions except no hemin was added, which obtained reduced graphene oxide (RGO).
Investigation of the peroxidase-like activity of GHs
Firstly, GHs solution (20 μL, 5 μg mL−1) was added in 60 μL PB buffer (125 mM, pH 5.0). Then 10 μL of ABTS (50 mM) and 10 μL of H2O2 (100 mM) were added, so was the substrate of TMB (10 mM) and OPD (10 mM). Kinetic measurements were carried out immediately by monitoring the absorbance at 420 nm, 652 nm and 450 nm accordingly on a Shimadzu UV-2450 UV-Vis spectrophotometer. In contrast, the same color reaction was performed in the presence of reduced graphene oxide (RGO).Response to ssDNA of GHs
Different concentrations of P1 (10 μL) and the GH solution (10 μL, 10 μg mL−1) were mixed in 60 μL of PB buffer (125 mM, pH 5.0) incubating for 10 min. Then 10 μL of ABTS (10 mM) and 10 μL of H2O2 (200 mM) were added. Kinetic measurements were carried out immediately by monitoring the absorbance at 420 nm on a Shimadzu UV-2450 UV-Vis spectrophotometer.Ability to distinguish ssDNA and dsDNA of GHs
P1 was hybridized with T1 through annealing in Tris–HCl buffer (10 mM, pH 7.9) to obtain 2 μM dsDNA. Then P1 (2 μM, 10 μL) and the obtained dsDNA (2 μM, 10 μL) were respectively mixed with the GH solution (10 μL, 10 μg mL−1) in 60 μL PB buffer (125 mM, pH 5.0) incubating for 10 min. Finally, 10 μL of ABTS (10 mM) and 10 μL of H2O2 (200 mM) were added. Kinetic measurements were carried out as above.Label-free colorimetric assay based on GHs for DNA
The P1 (2 μM, 10 μL) was mixed with 10 μL of target DNA (T1) in 50 μL of buffer solution (50 mM NaH2PO4, pH 5.0) at 37 °C for 1 h; then 10 μL of GHs (10 μg mL−1) was added into the reaction buffer incubating for 10 min; finally 10 μL of ABTS (10 mM) and 10 μL of H2O2 (200 mM) were added followed immediately by kinetic measurements.Label-free colorimetric assay based on GHs for cocaine
The assay procedures could be described as follows: the cocaine aptamer P2 (500 nM, 10 μL) was mixed with 10 μL of cocaine sample in 50 μL of buffer solution (50 mM NaH2PO4, pH 3.6) at 37 °C for 1 h; then 10 μL of GHs (10 μg mL−1) was added into the reaction buffer incubating for 10 min; finally 10 μL of ABTS (5 mM) and 10 μL of H2O2 (100 mM) were added followed immediately by kinetic measurements.Results and discussion
Characterization of GHs
We prepared the graphene–hemin nanosheets through π–π interactions. This nanocomposite could be obtained by mixing the graphene oxide suspension with hemin using hydrazine and ammonia reduction as illustrated in Scheme 1. The preparation of GHs was characterized by UV-Vis spectra. The GO dispersion displayed a maximum absorption at 227 nm corresponding to the π–π* transition of an aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and a shoulder at 290–300 nm, which was attributed to the n–π* transition of the CO bond.29 The spectrum of the hemin solution exhibited a strong peak at 388 nm attributed to the Soret band of porphyrin. Once reduced, the GHs contained two characteristic absorption peaks at 265 nm and 418 nm. The absorption peak at about 265 nm corresponded to the reduced graphene oxygen (RGO), which was 38 nm red shifted compared to GO, while the characteristic absorption at 418 nm was attributed to the Soret band of hemin with a 30 nm red shift. Remarkably, the graphene dispersion without hemin functionalisation only displayed an absorption peak at 264 nm and the shoulder peak at 290–300 nm correlating to the CO bond disappeared. These observations indicated the occurrence of strong π–π stacking interactions between the RGO and hemin, resulting in the formation of GHs. In addition, the GH dispersion solution was stable and no precipitation was observed after being kept for several months (inset d in Fig. 1), while the graphene dispersion without hemin congregated quickly (inset c in Fig. 1). These results demonstrated that hemin molecules could protect the reduced graphene oxide to make it stable.
C bond and a shoulder at 290–300 nm, which was attributed to the n–π* transition of the CO bond.29 The spectrum of the hemin solution exhibited a strong peak at 388 nm attributed to the Soret band of porphyrin. Once reduced, the GHs contained two characteristic absorption peaks at 265 nm and 418 nm. The absorption peak at about 265 nm corresponded to the reduced graphene oxygen (RGO), which was 38 nm red shifted compared to GO, while the characteristic absorption at 418 nm was attributed to the Soret band of hemin with a 30 nm red shift. Remarkably, the graphene dispersion without hemin functionalisation only displayed an absorption peak at 264 nm and the shoulder peak at 290–300 nm correlating to the CO bond disappeared. These observations indicated the occurrence of strong π–π stacking interactions between the RGO and hemin, resulting in the formation of GHs. In addition, the GH dispersion solution was stable and no precipitation was observed after being kept for several months (inset d in Fig. 1), while the graphene dispersion without hemin congregated quickly (inset c in Fig. 1). These results demonstrated that hemin molecules could protect the reduced graphene oxide to make it stable.
 | ||
| Scheme 1 The preparation and response to DNA of GHs. | ||
 | ||
| Fig. 1 UV-Vis spectra of GO suspension (a), hemin solution (b), RGO suspension (c) and GHs suspension (d). Inset: photographs of GO (0.5 mg mL−1) (a), hemin (0.5 mg mL−1) (b), RGO (0.25 mg mL−1) (c) and GHs (200 μg mL−1) (d). | ||
AFM was also used to observe the morphology and thickness of GH. The thickness of RGO was about 0.69 nm (Fig. 2A and C) corresponding to previously reported single-layer exfoliated sheets.30 While the thickness of GH was about 1.19 nm (Fig. 2B and D), which showed a 0.5 nm increase in height after functioning with hemin. This was in agreement with the previous report and the GHs were deemed to have been obtained, accordingly.1 The formation of GHs was also characterized by TEM. Fig. S1A† indicated that there was nothing attached to the graphene nanosheet. However, the nanosheet being covered with small particles was obviously observed in S1B,† these were regarded as hemin. Considering the AFM and TEM data, we assumed that the reduced graphene nanosheets were covered by a monolayer of hemin.
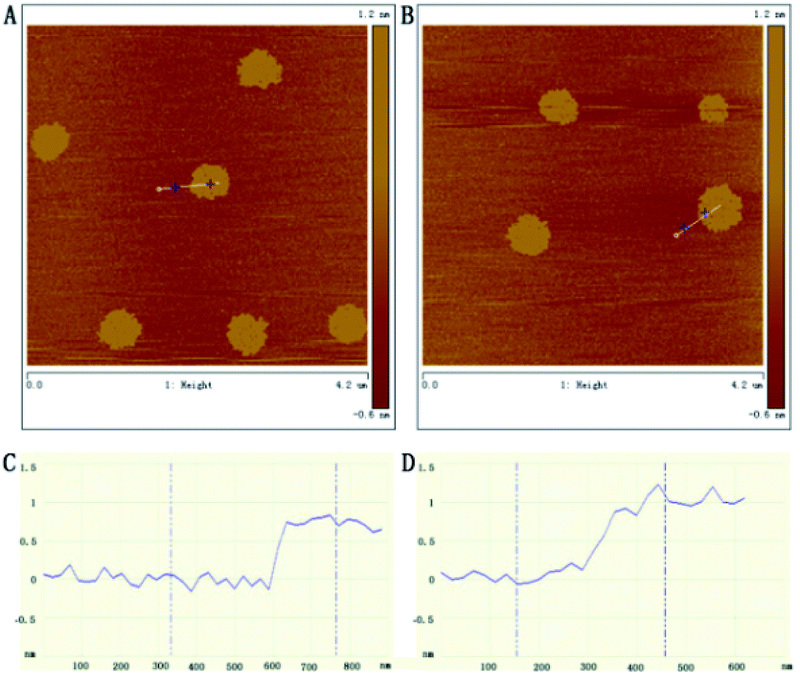 | ||
| Fig. 2 AFM images and height profiles of RGO (A, C) and GH (B, D). | ||
Peroxidase-like activity of GHs
It is well known that hemin is the activate site of the peroxidase enzyme and it exhibits peroxidase-like activity in a similar way to the peroxidase enzyme. Hence the GHs were supposed to have peroxidase-like activity. We investigated the catalytic oxidation reaction of GHs and RGO in the presence of different peroxidase substrates such as ABTS, TMB, and OPD; in the presence of H2O2, the color change was monitored by absorbance changes at 420 nm (ABTS), 652 nm (TMB) and 450 nm (OPD), respectively. As shown in Fig. S2A,† the GHs (b) catalyzed a large absorbance change, while only a slight change was observed in the presence of RGO (a). Similar phenomena were observed when selecting both TMB and OPD as substrates, as illustrated in Fig. S2B and S2C.† These observations indicated that GHs exhibited peroxidase-like activity while the activity of RGO was much less, demonstrating that hemin served as a catalyst similar to the peroxidase enzyme.Response to ssDNA and distinguishing ssDNA from dsDNA
For GHs, we found that they have the ability of responding to ssDNA in the presence of ABTS and H2O2 as shown in Scheme 1. It was observed that when P1 was mixed with the GHs, the colorimetric signal intensity increased significantly. Fig. 3A shows that the GHs mixed with 200 nM P1 displayed much higher peroxidase-like activity than GHs without P1, even with a low concentration of substrate and a buffer with an unsuitable pH. As the concentration of P1 increased, the oxidation product of ABTS showed a higher absorbance signal as illustrated in Fig. 3A. Previous reports demonstrated that many aromatic compounds can be stacked on DNA to some extent via π–π interactions.31,32 Therefore, the mechanism of this phenomenon was presumed to be that ssDNAs absorbed by hemin–rGO hybrid nanosheets have the ability of stacking ABTS and increasing its local concentration on the surface of hemin–rGO, which can, thus, lead to improved catalytic efficiency of hemin–rGO as well as the enhanced UV-Vis signal of the reaction system. To confirm this hypothesis, we further investigated whether the GHs could distinguish between ssDNA and dsDNA. Fig. 3B shows that GHs mixed with the hybrid of P1 and T1 had lower catalytic activity than those mixed with P1, which indicated that GHs had the ability of distinguishing ssDNA from dsDNA directly. Compared with traditional strategies associated with graphene, this phenomenon is novel and makes the as-prepared GHs an ideal nanocomposite to use in colorimetric sensors. | ||
| Fig. 3 (A) Time-dependent absorbance changes at 420 nm in the presence of different amounts of P1 ranging from 0 to 200 nM (0, 10, 25, 30, 40, 50, 100, 200 nM). (B) Time-dependent absorbance changes at 420 nm in the presence of 200 nM ssDNA and dsDNA. | ||
GH-based biosensor for DNA assay
Based on this unique property of GHs, we assumed that this material could be applied in the detection of DNA. It is well known that sensitive detection of nucleic acids has widespread applications in gene expression and clinical disease diagnostics.33,34 Thus, strategies for sensitive and selective detection of DNA are urgently needed. Fig. 4A illustrates the analytical principle of a biosensor for a DNA assay. First, complementary DNA (P1) of the target DNA (T1) was introduced as a probe. In the absence of target DNA, the catalytic activity and signal intensity would be high owing to the existence of ssDNA. When the target DNA is added, the ssDNA turns into dsDNA through hybridization and the absorbance signal becomes significantly lower. | ||
| Fig. 4 (A) Schematic illustration of procedures for DNA detection. (B) Time-dependent absorbance changes at 420 nm in the presence of different amounts of target DNA (0, 10, 20, 30, 50, 200 nM). (C) Calibration curve corresponding to the absorbance for varying concentrations of target DNA with an intervening time of 10 min. Inset shows a linear calibration curve from 10 to 50 nM. The error bars represent the standard deviation of three experiments. | ||
Then we investigated the concentration dependence with the target DNA. The concentration of the probe was chosen as 200 nM. The concentrations of GHs, ABTS and H2O2 were 1 μg mL−1, 1 mM and 20 mM, respectively. Fig. 4B depicts kinetic spectroscopy in the presence of different amounts of target DNA (0, 10, 20, 30, 50, 200 nM). A gradual decrease in absorbance at 10 min was observed with an increasing concentration of the DNA target. Fig. 4C displays the relationship between the absorbance and different target DNA concentrations after an interval of 10 min. A linear relationship was observed between the absorbance and different target DNA concentrations with a correlation coefficient R2 = 0.9970. The calibration equation is A = 0.6736 − 0.0025C, where A is the absorbance and C refers to the DNA concentration. In terms of the 3σ rule, the detection limit was estimated to be 9 nM.
Furthermore, control experiments using T2 (single base mismatch), T3 (three base mismatch) or T4 (unrelated DNA) were performed to investigate the specificity of the proposed strategy. As illustrated in Fig. S3A,† the absorbance ratios were much lower (A0/A) in the presence of the non-complementary sequences compared with the target one, implying that the proposed sensor strategy had desirable selectivity and thus providing a simple but specific platform for DNA assays.
GH-based biosensor for small molecule assay
After observing that the colorimetric assay was effective for DNA, we supposed that a nucleic acid aptamer could be used as a DNA probe to detect corresponding targets such as proteins or small molecules. Due to the enormous impact of cocaine to human health, cocaine was chosen as the model analyte to develop the label-free colorimetric biosensor.The protocol of our method is shown in Fig. 5A. First, cocaine aptamer P2 was used as the probe. In the absence of the target, the mixture catalyzed a great absorption change of ABTS with the addition of H2O2. However, with added cocaine, the absorbance change greatly decreased. With the optimization of experimental conditions, we finally choose 50 nM as the probe concentration. The optimal pH was 3.6 and the concentrations of GHs, ABTS and H2O2 were 1 μg mL−1, 0.5 mM and 10 mM, respectively. As shown in Fig. 5B, in the absence of cocaine, the absorbance at 420 nm was almost twice as much as that with 50 mM of cocaine present, after 10 minutes of reaction, which demonstrated the feasibility of the proposal.
 | ||
| Fig. 5 (A) Schematic illustration of procedures for cocaine detection. (B) Time-dependent absorbance changes at 420 nm in the absence of cocaine (a) and in the presence of cocaine (b, 50 mM). | ||
Next, the effect of concentration of the target cocaine was investigated. Fig. 6A displays time-dependent absorbance changes in the presence of different amounts of target cocaine. As shown in Fig. 6A, a gradual decrease in absorbance at 10 min was observed with an increasing concentration of cocaine and a dynamic range from 0 to 50 mM. Fig. 6B depicts the relationship between the absorbance and different cocaine concentrations after an interval of 10 min. The assay exhibited a linear correlation in the concentration range of 500 μM to 5 mM. The calibration equation is A = 1.112 − 0.04978C, where A is the absorbance and C refers to the cocaine concentration. The corresponding correlation coefficient of the calibration curve is 0.9922. The detection limit was estimated to be 230 μM according to the 3σ rule.
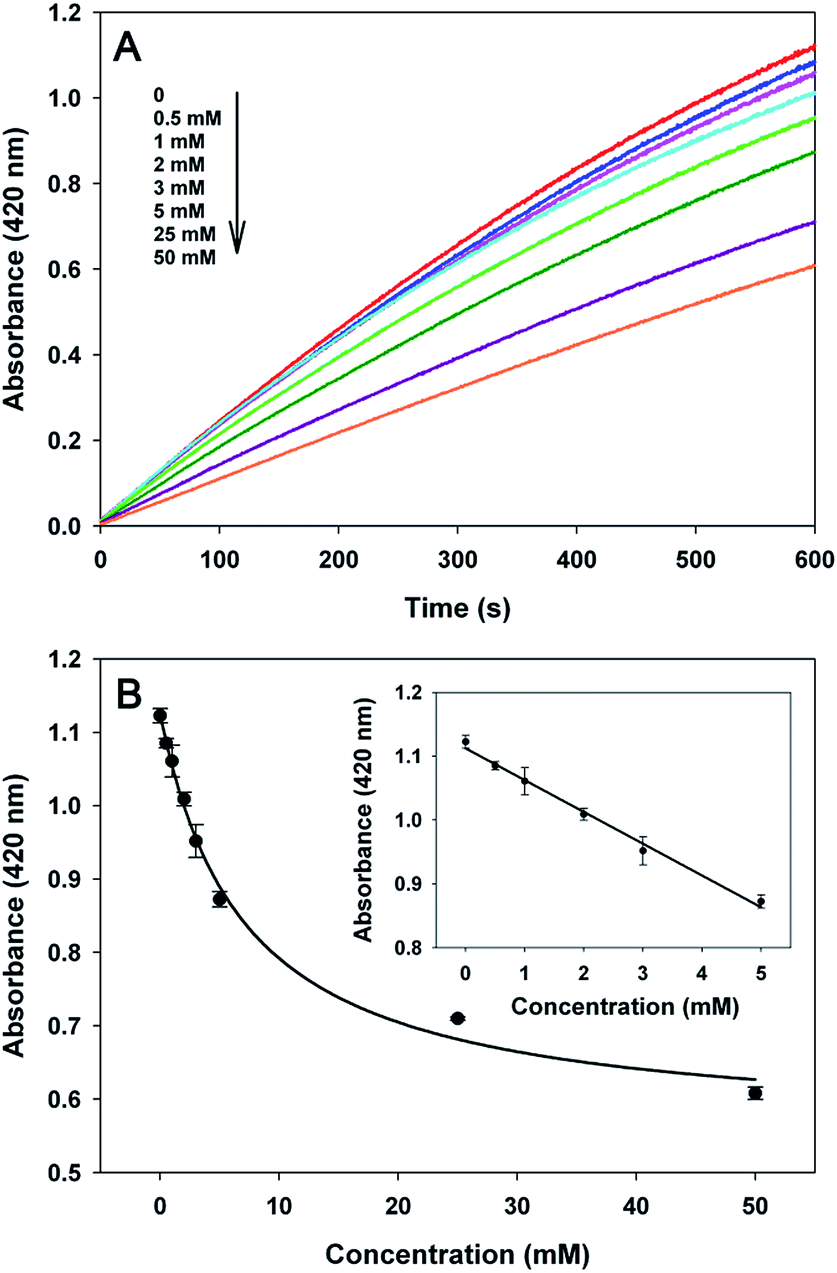 | ||
| Fig. 6 (A) Time-dependent absorbance changes at 420 nm in the presence of different amounts of cocaine ranging from 0 to 50 mM (0, 0.5, 1, 2, 3, 5, 25, 50 mM). (B) Calibration curve corresponding to the absorbance for various concentrations of cocaine with an intervening time of 10 min. Inset shows a linear calibration curve from 0.5 to 5 mM. The error bars represent the standard deviation of three experiments. | ||
Further investigation of the specificity of the sensor was performed. Three typical interfering compounds were chosen as controls: caffeine, theophylline and morphine hydrochloride (MHC). As illustrated in Fig. S4A,† nearly coincident kinetic data were observed with the addition of caffeine, theophylline and MHC (5 mM) compared with the blank, while the absorbance was decreased to a certain degree after a 10 min reaction following the addition of cocaine (5 mM). This result demonstrated that the biosensor had selectivity for cocaine. Additionally, the developed biosensor was able to detect other analytes by employing additional aptamers.
Conclusion
In summary, we have prepared hemin–graphene hybrid nanosheets which possess desirable water-solubility and stability. Especially, it is found that the GHs have peroxidase-like activity and the ability to identify ssDNA and dsDNA. Based on the special properties of the GHs, we have proposed a universal label-free colorimetric sensing strategy for DNA and cocaine assays. The proposed method showed advantages including operation simplicity, low cost and label-free design. By changing the sequence of DNA or using different aptamers, the proposed strategy has potential to be a universal platform for the detection of various analytes including protein, DNA and small molecules.Acknowledgements
This work was supported by NSFC (21025521, 21205034, 21035001, 21190041, 91317312), Doctoral Fund of Ministry of Education of China (New Teachers, 20120161120032), Hunan Provincial NSFC (13JJ4031). We also thank Meng-Meng Guo for her contribution in correcting the language of the manuscript.Notes and references
- Y. J. Guo, L. Deng, J. Li, S. J. Guo, E. K. Wang and S. J. Dong, ACS Nano, 2011, 5, 1282 CrossRef CAS PubMed.
- Y. Tao, Y. H. Lin, J. S. Ren and X. G. Qu, Biomaterials, 2013, 34, 4810 CrossRef CAS PubMed.
- T. Xue, S. Jiang, Y. Q. Qu, Q. Su, R. Cheng, S. Dubin, C. Y. Chiu, R. Kaner, Y. Huang and X. F. Duan, Angew. Chem., Int. Ed., 2012, 51, 3822 CrossRef CAS PubMed.
- B. Lin, Q. Q. Sun, K. Liu, D. Q. Lu, Y. Fu, Z. A. Xu and W. Zhang, Langmuir, 2014, 30, 2144 CrossRef CAS PubMed.
- W. Wei, D. M. Zhang, L. H. Yin, Y. P. Pu and S. Q. Liu, Spectrochim. Acta, Part A, 2013, 106, 163 CrossRef CAS PubMed.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752 CrossRef CAS PubMed.
- Y. X. Xu, H. Bai, G. W. Lu, C. Li and G. Q. Shi, J. Am. Chem. Soc., 2008, 130, 5856 CrossRef CAS PubMed.
- S. J. Liu, Q. Wen, L. J. Tang and J. H. Jiang, Anal. Chem., 2012, 84, 5944 CrossRef CAS PubMed.
- X. Q. Liu, R. Aizen, R. Freeman, O. Yehezkeli and I. Willner, ACS Nano, 2012, 6, 3553 CrossRef CAS PubMed.
- J. Zhang, M. J. Tao and Y. Jin, Analyst, 2014, 139, 3455 RSC.
- Z. S. Qian, X. Y. Shan, L. J. Chai, J. J. Ma, J. R. Chen and H. Feng, Nanoscale, 2014, 6, 5671 RSC.
- G. F. Zhang and P. K. Dasgupta, Anal. Chem., 1992, 64, 517 CrossRef CAS.
- Y. Xiao, V. Pavlov, T. Niazov, A. Dishon, M. Kotler and I. Willner, J. Am. Chem. Soc., 2004, 126, 7430 CrossRef CAS PubMed.
- R. Sun, Y. Wang, Y. N. Ni and S. Kokot, J. Hazard. Mater., 2014, 266, 60 CrossRef CAS PubMed.
- Y. Zhang, Z. Xia, H. Liu, M. J. Yang, L. L. Lin and Q. Z. Li, Sens. Actuators, B, 2013, 188, 496 CrossRef CAS PubMed.
- S. Bi, T. T. Zhao, X. Q. Jia and P. He, Biosens. Bioelectron., 2014, 57, 110 CrossRef CAS PubMed.
- Y. J. Guo, J. Li and S. J. Dong, Sens. Actuators, B, 2011, 160, 295 CrossRef CAS PubMed.
- Y. J. Li, X. Q. Huang, Y. J. Li, Y. X. Xu, Y. Wang, E. B. Zhu, X. F. Duan and Y. Huang, Sci. Rep., 2013, 3, 1787 Search PubMed.
- S. Y. Deng, J. P. Lei, Y. Huang, Y. Cheng and H. X. Ju, Anal. Chem., 2013, 85, 5390 CrossRef CAS PubMed.
- R. Frederick, H. Katherine and S. John, Analyst, 2009, 134, 701 RSC.
- F. Tagliaro, C. Poiesi, R. Aiello, R. Dorizzi, S. Ghielmi and M. Marigo, J. Chromatogr., 1993, 638, 303 CrossRef CAS.
- T. J. Beveridge, H. R. Smith, S. H. Nader, M. A. Nader and L. J. Porrino, Neuropharmacology, 2014, 85, 528 CrossRef CAS PubMed.
- J. Zhang, L. H. Wang, D. Pan, S. P. Song, F. C. Boey, H. Zhang and C. H. Fan, Small, 2008, 4, 1196 CrossRef CAS PubMed.
- J. L. He, Z. S. Wu, H. Zhou, H. Q. Wang, J. H. Jiang, G. L. Shen and R. Q. Yu, Anal. Chem., 2010, 82, 1358 CrossRef CAS PubMed.
- C. P. Ma, W. S. Wang, Q. Yang, C. Shi and L. J. Cao, Biosens. Bioelectron., 2011, 26, 3309 CrossRef CAS PubMed.
- A. V. Azaryan, L. J. Coughlin, B. Buzas, B. J. Clock and B. M. Cox, J. Neurochem., 1996, 66, 443 CrossRef CAS.
- L. Qiu, H. Zhou, W. P. Zhu, L. P. Qiu, J. H. Jiang, G. L. Shen and R. Q. Yu, New J. Chem., 2013, 37, 3998 RSC.
- K. Zhang, K. Wang, X. Zhu, J. Zhang, L. Xu, B. Huang and M. H. Xie, Chem. Commun., 2014, 50, 180 RSC.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101 CrossRef CAS PubMed.
- F. Liu and T. S. Seo, Adv. Funct. Mater., 2010, 20, 1930 CrossRef CAS.
- A. H. Christopher and K. M. S. Jeremy, J. Am. Chem. Soc., 1990, 112, 5525 CrossRef.
- P. G. W. Laurence, Med. Res. Rev., 1986, 6, 275 CrossRef.
- J. R. Lupski, J. G. Reid, C. Gonzaga-Jauregui, D. R. Deiros, D. C. Y. Chen and L. Nazareth, et al., N. Engl. J. Med., 2010, 362, 1181 CrossRef CAS PubMed.
- D. T. Miller, Y. Shen, L. A. Weiss, J. Korn, I. Anselm, C. Bridgemohan and G. F. Cox, et al., J. Med. Genet., 2009, 46, 242 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra08789e |
| This journal is © The Royal Society of Chemistry 2014 |