
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, biological evaluation and in silico and in vitro mode-of-action analysis of novel dihydropyrimidones targeting PPAR-γ†
Hanumantharayappa
Bharathkumar
a,
Shardul
Paricharak
bc,
K. R.
Dinesh
a,
Kodappully Sivaraman
Siveen
d,
Julian E.
Fuchs
b,
Shobith
Rangappa
ef,
C. D.
Mohan
g,
Nima
Mohandas
d,
Alan Prem
Kumar
dfhi,
Gautam
Sethi
d,
Andreas
Bender
*b,
Basappa
*a and
K. S.
Rangappa
*g
aLaboratory of Chemical Biology, Department of Chemistry, Bangalore University, Palace Road, Bangalore 560001, India. E-mail: salundibasappa@gmail.com
bUnilever Centre for Molecular Science Informatics, Department of Chemistry, University of Cambridge, Lensfield Road, CB2 1EW, Cambridge, UK
cDivision of Medicinal Chemistry, Leiden Academic Centre for Drug Research, Leiden University, P.O. Box 9502, 2300 RA Leiden, The Netherlands
dDepartment of Pharmacology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 117597, Singapore
eInterdisciplinary Research Group of Infectious Diseases, Singapore-MIT Alliance for Research & Technology Centre (SMART), 138602, Singapore
fCancer Science Institute of Singapore, National University of Singapore, Singapore 117599, Singapore
gDepartment of Chemistry, University of Mysore, Manasagangotri, Mysore 560001, India
hSchool of Biomedical Sciences, Faculty of Health Sciences, Curtin University, Western Australia, Australia
iDepartment of Biological Sciences, University of North Texas, Denton, Texas, USA
First published on 2nd September 2014
Abstract
Hepatocellular carcinoma, a fatal liver cancer, affects 600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people annually and ranks third in cancer-related lethality. In this work we report the synthesis and related biological activity of novel dihydropyrimidones. Among the tested compounds, 5-acetyl-4-(1H-indol-3-yl)-6-methyl-3,4-dihydropyrimidin-2(1H)-one (4g) was found to be most active towards the HepG2 cell line (IC50 = 17.9 μM), being at the same time 7.6-fold selective over normal (LO2) liver cells (IC50 = 136.9 μM). Subsequently, we identified peroxisome proliferator-activated receptor γ as a target of compound 4g using an in silico approach, and confirmed this mode-of-action experimentally.
000 people annually and ranks third in cancer-related lethality. In this work we report the synthesis and related biological activity of novel dihydropyrimidones. Among the tested compounds, 5-acetyl-4-(1H-indol-3-yl)-6-methyl-3,4-dihydropyrimidin-2(1H)-one (4g) was found to be most active towards the HepG2 cell line (IC50 = 17.9 μM), being at the same time 7.6-fold selective over normal (LO2) liver cells (IC50 = 136.9 μM). Subsequently, we identified peroxisome proliferator-activated receptor γ as a target of compound 4g using an in silico approach, and confirmed this mode-of-action experimentally.
The peroxisome proliferator-activated receptor (PPAR) belongs to the nuclear hormone receptor superfamily of transcription factors that includes 48 human transcription factors. Its activity is regulated by direct binding of steroid and thyroid hormones, vitamins, lipid metabolites, and xenobiotics.1 PPARs heterodimerize with retinoid X receptors, which get activated and bind to specific response elements in the target DNA of various target genes,2,3 PPARs consist of three different isoforms, PPAR-α, PPAR-β/δ, and PPAR-γ. Due to their central function in a variety of physiological processes, PPARs are important targets in drug discovery.4 Small molecules frequently interact with more than a single PPAR isoform leading to unique profiles of their biological effects.5
In particular, PPAR-γ has been established as a key regulator for inflammation,6 proliferation,7 metabolism,8 and differentiation,9 and it is also upregulated by many tumor suppressor genes.10 PPAR-γ is overexpressed in several types of human cancers, including breast, colon, bladder, and prostate cancer, and it also induces apoptosis in several malignant cell lineages.11 Furthermore, PPAR-γ ligands such as 15d-PGJ2 as well as antidiabetic thiazolidinediones (TZDs) function as anti-proliferative and proapoptotic agents, suggesting that PPAR-γ could be a promising therapeutic target for the treatment of cancer.12–14 Non-thiazolidinedione derivatives, such as 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid, GW-7845, JTT-501, KRP-297, L-764406, MCC-555 and GW-0207 are further known synthetic ligands of PPAR-γ.15
On the other hand, the natural product ectoine, a tetrahydropyrimidine derivative, offers protection against the effects of ischemia-reperfusion injury in intestinal transplantation in vivo.16 (±)-Aplicyanin analogs bearing a pyrimidone moiety showed significant anti-tumor activity via targeting p38 MAP kinase.17–19
Similar small molecules, dihydropyrimidines (DHPs), have also been reported as anti-cancer agents.20 Recently, pyrimidine-tethered curcumins showed anticancer activity by targeting EGFR tyrosine kinase.21
In continuation of our interest to synthesize novel bioactive heteocycles,22–24 we herein report the synthesis of DHP tethered to various functional heterocycles like indole, flavone and benzofuran. Furthermore, and conceptually rather novel, we rationalized the mode-of-action for the lead DHP as PPAR-γ via an in silico cheminformatics approach, followed by experimental validation. Details on synthesis, compound chararacterization, biological assays, and in silico methods are described in more detail in the ESI.†
For synthesis, multi-component reactions (MCRs) have been employed which constitute an efficient synthetic strategy for rapid and effective laboratory organic transformations, because products are prepared in a one-pot and single step approach and the diversity can be obtained directly by changing the reacting components.25 Here, we synthesized the dihydropyrimidone bearing small molecules via multi-component Biginelli reaction.26 The mechanism for the formation of the title compounds involves the condensation between the aldehyde and urea to form an iminium intermediate, which acts as electrophile for the nucleophilic addition of the ketoester enol, and the ketone carbonyl of the resulting adduct undergoes condensation with the amine group of urea to give the cyclized product (Fig. 1A). Also further heterocycles such as indole, flavone and benzofuran moieties were successful employed for the preparation of novel dihydropyrimidones (see ESI Table 1†). The protocol was effective with aromatic amines having electron donating groups, and products were identified based on IR, LCMS, 1H NMR, and 13C NMR spectra (see ESI†). In the next step, the cytotoxic effects of dihydropyrimidones was investigated in HepG2 cell lines using the MTT assay. The cells were treated with 0, 10, 25, 50, and 100 μM concentrations of dihydropyrimidones for 72 h. DHPs were found to inhibit the viability of HepG2 cells in a dose-and time-dependent manner. Compound 4g, 4k and 4p were found to be most effective with IC50 values less than 50 μM. In addition, we were able to identify that all the dihydropyrimidone series of compounds inhibited the viability of immortalized normal human liver cells, LO2, at higher concentrations. Compound 4g was found have an IC50 value of 17.9 μM against HepG2 and has the highest, 7.6-fold selectivity over the normal LO2 liver cell line (IC50 = 136.93 μM; see ESI Table 2† for more details). Therefore, we considered 4g (Fig. 1B) in more detail and studied its mode-of-action in HepG2 cells.
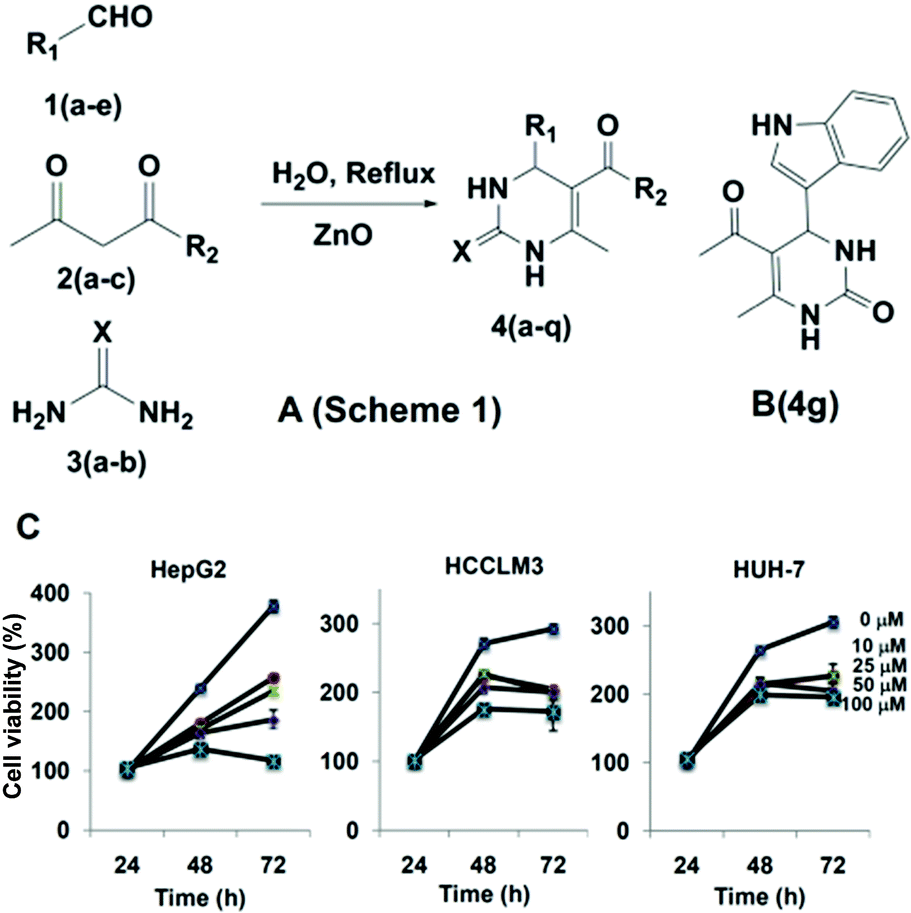 | ||
| Fig. 1 Synthetic scheme of novel dihydropyrimidones and cytotoxicity studies for the lead compound 4g against various HCC cells. | ||
Human protein targets were predicted for the most bioactive compound 4g, and of all predicted targets (ESI Table 3†), PPAR-γ was the only target with score of 26.02, exceeding the significance cut-off of 10. Given the involvement of PPAR-γ in the induction of apoptosis and cancer development, as described in previous studies,4–6 this target was selected for further analysis.
PPAR-γ has been extensively shown to be associated with anti-cancer effects in a variety of cancer types including HCC. HepG2 cells were treated with different concentrations of compound 4g for 8 h and then analyzed for the expression of PPAR-γ by western blot analysis. It was found that compound 4g could substantially increase PPAR-γ expression in a dose-dependent manner, with maximum effect at 15 μM concentration (Fig. 2A). Compound 4g also increased PPAR-γ expression in a time-dependent manner, with maximum activity at 8 h (Fig. 2B). Evidently, the DNA-binding assay for PPAR-γ in nuclear extracts showed that compound 4g significantly enhanced PPAR-γ DNA binding ability in a time dependent manner (Fig. 2C).
 | ||
| Fig. 2 In silico and in vitro rationalization of the mode-of-action analysis for the lead compound 4g that targets PPAR-γ in HepG2 cells. | ||
DHPs are well known ligands for PPAR-γ. In order to determine the interaction of dihydropyrimidones towards PPAR-γ, the co-crystal structure of human PPAR-γ in complex with rosiglitazone was considered for our studies.27 Using Discovery Studio (DS) default tools and settings, the ligand binding domain (LBD) of PPAR-γ was identified and ligands were prepared for docking utilizing the CDOCKER programme of the ligand–receptor interaction module of Discovery Studio version 2.5, the dihydropyrimidones were docked into the LBD of PPAR. The CDOCKER energies for all docked ligands (ESI Table 4†) indicate that compound 4g bound to the LBD region with the lowest (and thus most favorable) score of −24.0 kcal mol−1 (ESI Table 3†). Furthermore, the binding pose of compound 4g was found to overlap with the hydrophobic tail of TZDs (rosiglitazone; Fig. 2D). Compound 4g forms mainly hydrophobic contacts with residues at the LBD region of PPAR-γ including Ile281, Met348, Ile353, Val339, Cys285, Ile341, Ser342, Gly284, Tyr321, Ile326, Lys367, Phe363, Met364, Leu330, Ser289, and Arg288 (Fig. 2E). Thereby, CH–π interactions with Leu-330 and van der Waals contacts with the alkyl chain of Arg-288 are proposed as main driving forces of the ligand. This molecular docking study revealed that compound 4g could be a suitable non-TZD ligand for PPAR-γ.
We investigated whether activation of PPAR-γ in HepG2 cells by compound 4g leads to apoptosis. In HepG2 cells treated with compound 4g, there was a time-dependent reduction in the levels of procaspase-3 (Fig. 3A). This suggests that cleavage events had occurred, indicating the activation of caspase-3, the levels of which were shown to be increased after 48 h. Activation of caspase-3 led to the cleavage of a 118 kDa Poly (ADP-ribose) polymerase (PARP) protein into an 85 kDa fragment (Fig. 3B). This result clearly suggests that compound 4g induces caspase-3-dependent apoptosis in HepG2 cells. In addition, compound 4g was found to downregulate the anti-apoptotic and proliferative proteins, Bcl-2 and Cyclin D1 in a time dependent manner (Fig. 3C), with maximum effect observed at 48 h. Pronounced expression of PPAR-γ was demonstrated in HCC cells treated with rosiglitazone and such induction markedly suppressed the migration of HCC cells.28 The effect of compound 4g on the migration of HepG2 cells was investigated using a wound healing assay. We found that compound 4g treatment significantly suppressed the migration of HepG2 cells, and the pretreatment with GW9662, a pharmacological PPAR-γ inhibitor, reversed the anti-migratory effects of compound 4g as shown in Fig. 3D and E. It is well known that PPAR-γ ligands may have therapeutic value for the treatment of highly invasive breast cancer by targeting invasion.29 An in vitro Bio-Coat Matrigel invasion assay was performed (BD Biosciences, San Jose, CA, USA), and it was found that upon treatment with compound 4g there was a significant reduction in the number of cells that could invade the Matrigel coated polycarbonate membrane, indicating that compound 4g could indeed significantly inhibit the invasive property of HepG2 cells (Fig. 3F and G). Moreover, we found that pretreatment with the PPAR-γ antagonist GW9662 reversed the anti-invasive potential of compound 4g in HCC cells, confirming that the activity is mediated through inhibition of PPAR-γ pathway.
 | ||
| Fig. 3 Apoptotic induction, anti-migration, and anti-invasive activity of the compound 4g in HCC cells. | ||
In conclusion, we herein report the synthesis of novel dihydropyrimidones and found 5-acetyl-4-(1H-indol-3-yl)-6-methyl-3,4-dihydropyrimidin-2(1H)-one (4g) to be most active against HCC cells. Furthermore, compound 4g down-regulated anti-apoptotic and proliferative proteins such as Bcl-2 and Cyclin D1 in a time-dependent manner. In silico studies predicted PPAR-γ as mode-of-action of compound 4g, which could be validated by in vitro studies. Molecular docking furthermore suggested the binding pose of 4g within the ligand binding domain of PPAR-γ, which overlaps with the tail of rosiglitazone. Compound 4g inhibited the invasive property of HepG2 cells and the pretreatment with GW9662 reversed the anti-invasive potential of compound 4g in HCC cells in vitro, thereby confirming that the activity is mediated through inhibition of PPAR-γ pathway. Hence, both in silico and experimental methods agree on the mode-of-action of compound 4gvia PPAR-γ.
Acknowledgements
This research was supported by the Department of Science and Technology (no. SR/FT/LS-142/2012), University Grants Commission (41-257-2012-SR), and Vision Group Science and Technology to Basappa. Basappa thanks Karnataka University, INDIA for DC PAVATE Fellowship and IISc, Bangalore for providing access to the NMR Facility. Bharathkumar thanks UGC for a BSR fellowship. SP thanks the Netherlands Organisation for Scientific Research (NWO, grant number NWO-017.009-065) and the Prins Bernhard Cultuurfonds for funding. CDM thanks DST for INSPIRE fellowship.Notes and references
- M. Lehrke and M. A. Lazar, Cell, 2005, 123, 993–999 CrossRef CAS PubMed.
- F. S. Celi and A. R. Shuldiner, Curr. Diabetes Rep., 2002, 2, 179 CrossRef PubMed.
- C. Grommes, G. E. Landreth and M. T. Heneka, Lancet Oncol., 2004, 5, 419–429 CrossRef CAS.
- T. M. Willson, P. J. Brown, D. D. Sternbach and B. R. Henke, J. Med. Chem., 2000, 43, 527–550 CrossRef CAS.
- M. Dietz, P. Mohr, B. Kuhn, H. P. Maerki, P. Hartman, A. Ruf, J. Benz, U. Grether and M. B. Wright, ChemMedChem, 2012, 7, 1101–1111 CrossRef CAS PubMed.
- C. Jiang, A. T. Ting and B. Seed, Nature, 1998, 391, 82–86 CrossRef CAS PubMed.
- H. P. Koeffler, Clin. Cancer Res., 2003, 9, 1–9 CAS.
- J. M. Lehmann, L. B. Moore, T. A. Smith-Oliver, W. O. Wilkinson, T. M. Willson and S. A. Sliewer, J. Biol. Chem., 1995, 270, 12953–12956 CrossRef CAS PubMed.
- S. A. Kliewer, J. M. Lenhard, T. M. Willson, I. Patel, D. C. Morris and J. M. Lehmann, Cell, 1995, 83, 813–819 CrossRef CAS.
- L. Patel, I. Pass, P. Coxon, C. P. Downes, S. A. Smith and C. H. Macphee, Curr. Biol., 2001, 11, 764–768 CrossRef CAS.
- E. Elstner, C. Muller, K. Koshizuka, E. A. Williamson, D. Park, H. Asou, P. Shintaku, J. W. Said, D. Heber and H. P. Koeffler, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8806–8811 CrossRef CAS.
- G. Venkatachalam, A. P. Kumar, K. R. Sakharkar, S. Thangavel, M. V. Clement and M. K. Sakharkar, J. Drug Targeting, 2011, 19, 781–796 CrossRef CAS PubMed.
- M. Pignatelli, J. Sanchez-Rodriguez, A. Santos and A. Perez-Castillo, Carcinogenesis, 2005, 26, 81–92 CrossRef CAS PubMed.
- I. W. Campbell, Curr. Mol. Med., 2005, 5, 349–363 CrossRef CAS.
- E. H. Kim and Y. J. Surh, Biochem. Pharmacol., 2008, 76, 1544–1553 CrossRef CAS PubMed.
- T. Pech, I. Ohsawa, M. Praktiknjo, M. Overhaus, S. Wehner, M. von Websky, K. Abu-Elmagd, G. van Echten-Deckert, J. C. Kalff and N. Schaefer, Pathobiology, 2013, 80, 102–110 CrossRef CAS PubMed.
- M. Sisa, D. Pla, M. Altuna, A. Francesch, C. Cuevas, F. Albericio and M. Alvarez, J. Med. Chem., 2009, 52, 6217–6223 CrossRef CAS PubMed.
- T. Asano, H. Yamazaki, C. Kasahara, H. Kubota, T. Kontani, Y. Harayama, K. Ohno, H. Mizuhara, M. Yokomoto, K. Misumi, T. Kinoshita, M. Ohta and M. Takeuchi, J. Med. Chem., 2012, 55, 7772–7785 CrossRef CAS PubMed.
- S. Cesarini, A. Spallarossa, A. Ranise, S. Scenone, C. Rosano, P. L. Colla, G. Sanna, B. Busonera and R. Loddo, Eur. J. Med. Chem., 2009, 44, 1106–1118 CrossRef CAS PubMed.
- B. R. Prashantha Kumar, G. Senkar, R. B. Nasir Baig and S. Chandrashekaran, Eur. J. Med. Chem., 2009, 44, 4192–4198 CrossRef CAS PubMed.
- M. J. Ahsan, H. Khalilullah, S. Yasmin, S. S. Jadav and J. Govindasamy, BioMed Res. Int., 2013, 239354 Search PubMed.
- Basappa, M. P. Sadashiva, K. Mantelingu, S. N. Swamy and K. S. Rangappa, Bioorg. Med. Chem., 2003, 11, 4539–4544 CrossRef CAS PubMed.
- Basappa, S. Murugan, C. V. Kavitha, A. Purushothaman, K. G. Nevin, K. Sugahara and K. S. Rangappa, Cancer Lett., 2010, 297, 231–243 CrossRef CAS PubMed.
- Basappa, K. Sugahara, K. N. Thimmaiah, H. K. Bid and K. S. Rangappa, PLoS One, 2012, 7, e39444 CAS.
- P. Slobbe, E. Ruijter and R. V. A. Orru, Med. Chem. Commun., 2012, 3, 1189–1218 RSC.
- C. O. Kappe, Tetrahedron, 1993, 49, 6937–6963 CrossRef CAS.
- M. V. Liberato, A. S. Nascimento, S. D. Ayers, J. Z. Lin, A. Cvoro, R. L. Silveira, L. Martinez, P. C. Souza, d. Saidemberg, T. Deng, A. A. Amato, M. Togashi, W. A. Hsueh, K. Phillips, M. S. Palma, F. A. Neves, M. S. Skaf, P. Webb and I. Polykarpov, PLoS One, 2012, 7, e36297 CAS.
- B. Shen, E. S. Chu, G. Zhao, K. Man, C. W. Wu, J. T. Cheng, G. Li, Y. Nie, C. M. Lo, N. Teoh, G. C. Farrell, J. J. Sung and J. Yu, Br. J. Cancer, 2012, 106, 1486–1494 CrossRef CAS PubMed.
- H. Liu, C. Zang, M. H. Fenner, K. Possinger and E. Elstner, Breast Cancer Res. Treat., 2003, 79, 63–74 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical structures of all educts and products as well as synthesis protocols and spectroscopic characterization of the compounds are available as ESI. See DOI: 10.1039/c4ra08713e |
| This journal is © The Royal Society of Chemistry 2014 |