
Two stage chemical bath deposition of MoO3 nanorod films
Arpan Dharaa,
Gary Hodesb and
Shaibal K. Sarkar*a
aDepartment of Energy Science and Engineering, Indian Institute of Technology Bombay, Mumbai-400 076, India. E-mail: shaibal.sarkar@iitb.ac.in
bDepartment of Materials and Interfaces, Weizmann Institute of Science, Rehovot-76100, Israel
First published on 14th October 2014
Abstract
Thin films of hexagonal MoO3 nanorods are deposited by chemical bath deposition from a highly acidic aqueous solution containing ammonium heptamolybdate at 85 °C. We present a unique two-stage deposition process that results in uniform film formation even from a highly depleted solution. Growth occurs during the second stage only if the substrate removed from the first stage solution is first rinsed with water. Mechanistically, the proposed process is a combination of formation of nucleation sites at the first stage, removal or modification of a passivation layer on the nucleation sites by the rinse and chemical heterogeneity-induced growth at the second stage. Variations in either of these two deposition stages lead to control over rod dimensions and film texture. An FTIR study confirms the presence of amine and hydroxyl groups tightly bound in the crystals. Furthermore, h-MoO3 nanorod films showed good photocatalytic activity towards degradation of methylene blue (MB) under visible light.
1. Introduction
Transition metal oxides and sulfides with layered structures offer interesting applications in various fields ranging from electronics to mechanical lubrication.1–4 MoO3 in particular is suitable for a wide range of applications in electronics,2,5,6 photovoltaic energy conversion7,8 and electrochemical storage9,10 applications. The possibility of ion intercalation in layered structures of MoO3 make it highly pertinent in solid-state Li-ion batteries.11–13 The high work function and low-lying conduction band of MoO3 is responsible for its ability to facilitate hole removal in organic photovoltaics.14,15 Apart from its bulk material properties, its nanostructured forms provide an added parameter of high surface to volume ratio that results in higher catalytic,16–19 sensing20 and charge collection21 efficiencies.Several studies have described the synthesis of MoO3 nanostructures of various shapes and sizes. Orthorhombic α-MoO3, which is the thermodynamically most stable phase, has a unique layered atomic structure, but various other phases are found in the literature.22–24 Most of the reported syntheses involve high temperatures involving mostly gas phase synthesis in high vacuum. Vacuum arc synthesis is one among the many.25 Baker et al. described a solution phase synthesis of a high pressure phase of MoO3.26 In other studies a mixed phase of α-MoO3 and β-MoO3 (monoclinic) was obtained.22,23 A popular way to deposit films of MoO3 is through post-deposition thermal annealing of evaporated Mo metal films.27 Deposition of MoO3 nanorods or other nanostructures were reported but all are high energy process.28–30
Decomposition of ammonium heptamolybdate (HMA) to form thermodynamically stable α-MoO3 by a hydrothermal method is well documented. The reaction mechanism in solution can be written as follows:31
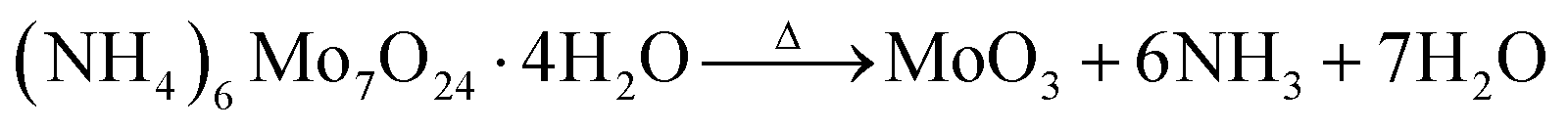 | (1) |
Control over the morphology and particle size can be further obtained by the addition of additives that can complex with the Mo6+. Song et al. reported a simple precipitation method to produce bulk precipitation of hexagonal-MoO3 (h-MoO3) nanorod structures by acidifying HMA.32 The isopolymolybdate ions (Mo7O246−) in the solution were converted to MoO3 in the presence of excess H+ ions. The reaction mechanism can be written as
| Mo7O24 + 6H+ = 7MoO3 + 3H2O. | (2) |
This opens a new arena to prepare and study the properties of meta-stable h-MoO3 that can be expected to behave differently than the thermodynamically-stable α-MoO3 phase. Precipitation of h-MoO3 by acidification of HMA was demonstrated in 1987 by Kumagai et al.33 However it is difficult to distinguish it from its other cation-stabilized form, MoO3·nNH3·mH2O22,34 due to the similarities in the XRD patterns. Apart from the simple precipitation techniques, h-MoO3 was also synthesized through hydrothermal22,35 and vapor deposition36 routes. Solution deposition of MoO3 thin films was much less studied than precipitates. High purity h-MoO3 films with good crystallinity were deposited by Deki et al. from a bath containing molybdic acid dissolved in a mixture of HF and boric acid.37
In this paper we describe conditions to obtain films of h-MoO3 by acidification of HMA. In this precipitation, no film formation occurred under most conditions and, where it did form, the film quality (coverage) was poor. Based on these results, we developed a unique two-stage deposition of good quality, oriented h-MoO3 nanorod films by CBD. In the first stage, only nucleation occurred on the fluorine-doped tin oxide (FTO)-coated glass substrate, followed by film growth during the second stage. Control over the rod length, diameter and orientation was achieved by controlling the deposition conditions.
2. Experimental
MoO3 films were deposited from a highly acidic bath containing 0.1–0.05 M of HMA and 5.2 M of HNO3 in a thermostatically-controlled water bath at 85 °C for 1 h. The deposition solution was placed in an airtight glass vial with the FTO-coated glass substrate placed facing down at a roughly 45° angle to the bottom of the vial to avoid accumulation of bulk precipitate on the substrate surface. After deposition, the surface was rinsed thoroughly with DI water before drying with N2 gas.For two-stage deposition, 5 ml of 2 M HNO3 was mixed with 10 ml HMA (concentration varied from 0.1–0.01 M) solution with continuous stirring. About 10 ml of that solution transferred to a vial and kept into a water bath at 85 °C. Then the FTO substrates were placed into the solution as described earlier for another half an hour. This step is considered as nucleation step. After that, substrates were taken out from the solution and ringed thoroughly with distilled water followed by the growth step where the substrates were again put into the same solution for another half an hour. After deposition, films were washed with distilled water and dried.
Photocatalytic degradation study of methylene blue (MB) dye using h-MoO3 nanorods films were performed in a quartz vessel containing 25 ml dye solution (10 mg L−1). Two thin films of h-MoO3 with active area 2.5 × 1 cm2 each were dipped into the dye solution and a tungsten halogen light of 300 W was used as a light source. UV-VIS spectroscopy of the solution as a function of the irradiation time was performed separately.
X-Ray diffraction measurements were performed in the θ–2θ configuration, using a Phillips X'Part diffractometer equipped with a Cu anode operating at 40 kV and 30 mA, emitting a wavelength of 1.54 Å.
Scanning Electron Microscope (SEM) images were taken with a JEOL FESEM using a secondary electron detector. For Transmission Electron Microscopy (TEM), films were scraped from the substrate with a blade and dispersed in water, then put on a grid and dried naturally. TEM imaging was carried out with a JEOL 2100F transmission electron microscope operating at 200 kV.
Fourier Transform Infra Red (FTIR) spectroscopy measurements were performed with a Bruker Vertex 70 in transmission mode. Samples were scraped from the film, heated where needed, and mixed well with KBr powder to form pellets.
Absorbance measurements were done with a PerkinElmer Lambda 35 UV-VIS spectrophotometer in absorption mode.
3. Results and discussions
Under highly acidic condition, heating of HMA results in precipitation of MoO3. In this process, isopolymolybdate ions (Mo7O246−) in the solution are converted to MoO3 upon heating at 85 °C (as shown in eqn (2)). X-ray diffraction and SEM imaging confirmed the hexagonal crystallographic nature of the powdery precipitate. Film formation was found only on the FTO surface: no film formation was observed on the glass container surface, activated glass (HNO3 or KMnO4 treated) or any metal substrates tried by us (stainless steel or Au). However we did find film growth occurred also on TiO2. We did not devote much effort to trying to understand what was special about the FTO and TiO2. We do note that, since the solution was highly acidic, all substrates are expected to be positively charged and therefore the reason is unlikely to be due mainly to electrostatic forces. All references to films in this paper refer to films on FTO.An XRD pattern of a representative as-deposited film is shown in Fig. 1a. All the peaks can be indexed to the hexagonal phase of MoO3. Crystallographic analysis of the pattern reveals a = b = 10.5756 Å and c = 3.7209 Å which matches with the Rietveld fitting value done by Pan et al.38 as well as with other literature values.34,37 An SEM image of the same film is shown in Fig. 1b. A rod-like morphology with rod diameter ranging from 2–10 μm and with flat hexagonal top surfaces is clearly seen. A TEM image of a single rod is shown in Fig. 1c together with selected point-like ED pattern, reveals the single crystalline nature of the individual rods.
 | ||
| Fig. 1 (a) XRD pattern of the as-deposited films obtained from 0.1 M HMA and 5.2 M HNO3 in the Bragg–Brentano configuration. (b) SEM micrograph revealing hexagonal rod-like morphology of the deposit. (c) TEM image and the SAED pattern showing mono crystallinity of the individual rods. | ||
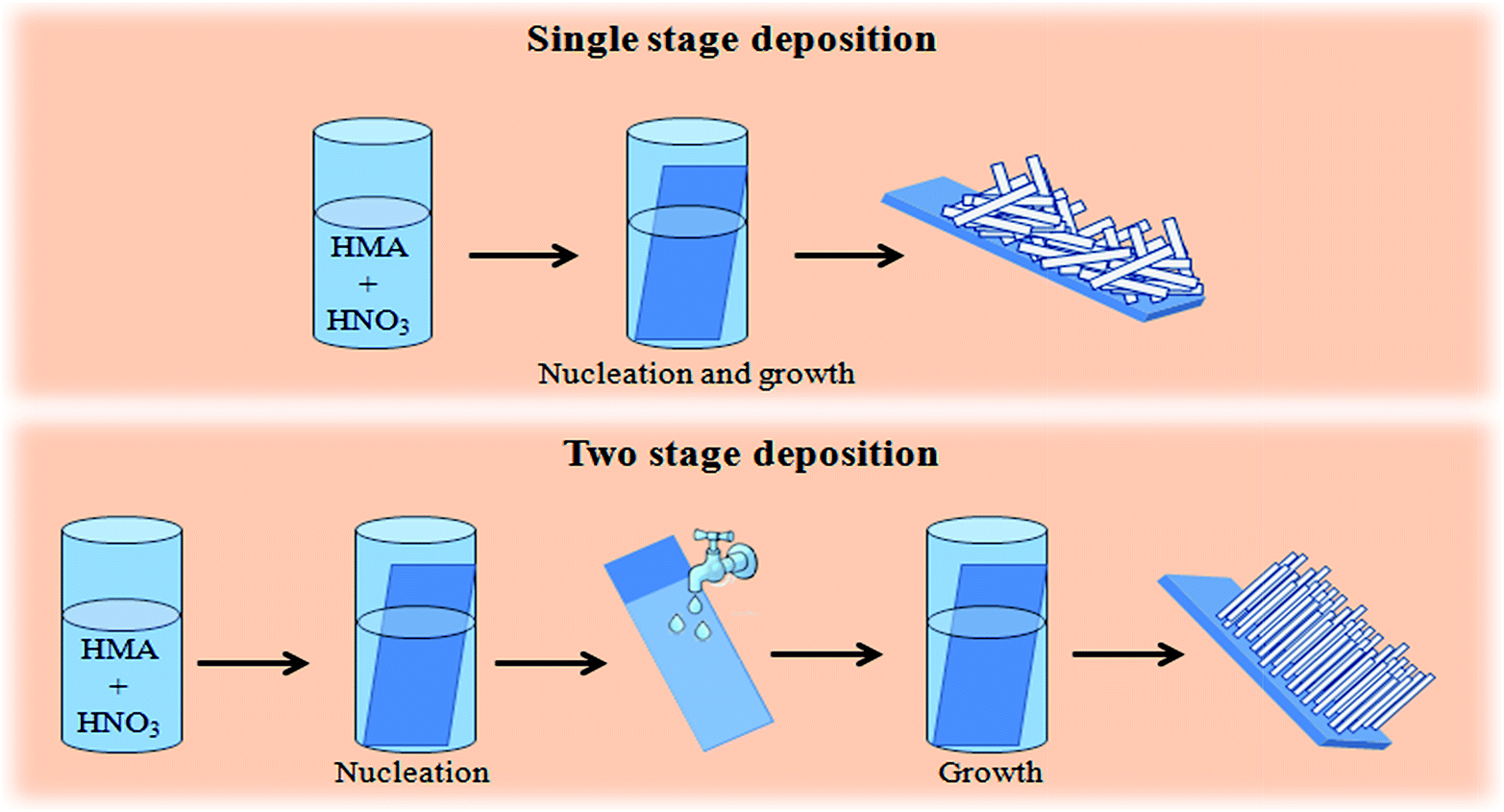 | ||
| Fig. 2 Schematic representation of single stage and double stage deposition process of h-MoO3 nanorod films. | ||
Semi-quantitative XPS elemental analysis of the as deposited films (Fig. 3a) shows Mo 3d5/2 and 3d3/2 peaks at 232.686 eV and 235.847 eV, agreeing with that reported previously for MoO3.32,39 However, a considerable amount of nitrogen at 398.6 eV was observed which is chemically different from the nitrogen in metal nitrides. To understand the chemical nature of the nitrogen in the deposit, we performed FTIR transmission measurements on powder scraped from the substrates. As shown in Fig. 3b, –NH bending and stretching peaks were clearly observed at ca. 1440 cm−1 and 3228 cm−1 respectively.34,40 Also absorbance from –OH vibrations was found at 1400, 1610 and 3500 cm−1. The peak at 1600 cm−1 corresponds to the deformation of free water molecule and that at 1410 is probably the MoO–H bending vibration.34 To understand the nature of the –OH and –NH peaks, samples were annealed at 200 °C. After this annealing, the –OH peak intensity did not change whereas the –NH peak intensity decreased by ca. 50%. The lack of change of the water/OH peaks on annealing suggests that these species are inside the crystals rather than predominantly on the surface, where they (at least the water) would be expected to be strongly reduced in concentration on annealing. The N most likely comes from NH4+ that is mainly also inside the crystal – either occluded or as MoO3·nNH3·mH2O.
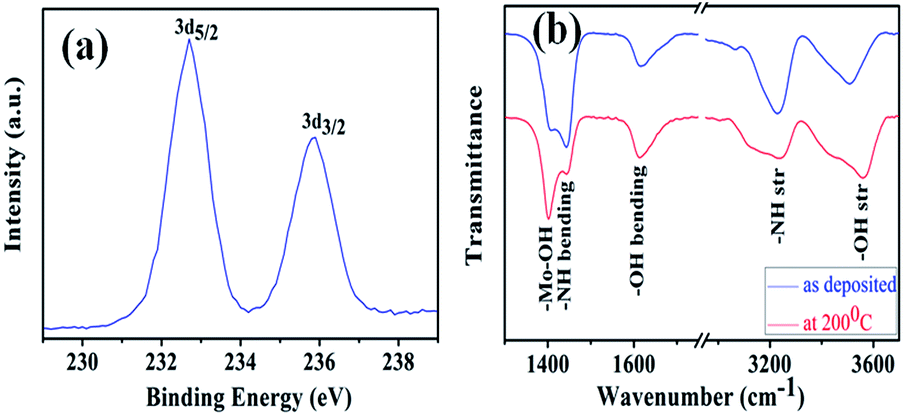 | ||
| Fig. 3 (a) XPS Mo peaks obtained from an as-deposited film (b) FTIR transmission spectra showing N–H and O–H stretching and bending vibrations of as-deposited and air-annealed (at 200 °C) samples. | ||
Reducing the relative HMA concentration from 0.1 M to 0.05 M, while keeping the HNO3 concentration unaltered (5.2 M), resulted in a decrease of both the rod diameter and the surface coverage as shown in Fig. 4(a–c).While the rod formation can be attributed to the higher reactivity of the (101) surface, the low material concentration resulted in poor surface coverage. Low ionic concentration favors lower nucleation density on the substrate. Similar behavior was observed during ZnO growth in the presence of relatively low concentrations of Zn2+.41
 | ||
| Fig. 4 Scanning electron micrograph of the films obtained with (a) 0.1 M, (b) 0.065 M and (c) 0.05 M HMA concentration. The HNO3 concentration was constant at 5.2 M. | ||
The film formation largely depends on the acid concentration in the deposition solution. No visible deposition occurs if the acid concentration is reduced below 5.2 M, other deposition parameters remaining unaltered, although bulk precipitation occurs in the solution even at much lower acid concentration (>0.05 M). The rate of this precipitation depends on the net acid concentration in the solution. After addition of the acid to the HMA solution at 85 °C, the transparent solution slowly turns increasingly turbid, then gradually transparent again as the suspension precipitates. To exemplify the dependence of these processes on HNO3 concentration, for 5.2 M HNO3 with 0.1 M HMA, it takes one min for the solution to become cloudy and another10 min to clear, indicating the completion of bulk precipitation. Reducing the acid concentration, it requires more time for the solution to become turbid and the turbidity persists for a longer time. Thus reduction in acid concentration in the deposition solution lowers the reaction rate. The above-described behavior also clearly indicates that homogeneous nucleation occurs in the bulk of the solution, resulting in a suspension that finally precipitates. In parallel with homogeneous nucleation and precipitation, nucleation and growth obviously also occurs on the FTO substrate (but not on most other substrates we tried).
However, when a substrate that was removed from the reaction solution after 30 min and showed no visible deposit, was rinsed with DI water and then again immersed in the same (or a fresh) solution, a highly uniform film was deposited. In this paper, we identify such a film as a second stage deposition, while the initial 30 min treatment is identified as a first stage deposition (Fig. 2). The bulk precipitation that occurred during the first stage of the two-stage reaction considerably lowered the molybdenum concentration in the solution. Before describing these deposits, since it is clear that, although no visible deposit formed after the first stage, there is some effect of this first stage treatment, most likely nucleation of some species, we investigated more closely this first stage. We studied the substrate after 30 min in the (5.2 M) deposition solution.
While FESEM images revealed no distinct particles that might be attributed to nuclei, XPS clearly indicated the presence of Mo+6 species on the substrate (Fig. 5). The ratio of surface Mo+6 to Sn+4 (from the FTO substrate) after the first stage was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4.25. Assuming the Mo is in the form of discrete nuclei of a Mo–O material (the most likely expected scenario), this would represent a moderately dense coverage of nuclei with comparable diameter and spacing between the nuclei. Taking into account the relatively close atomic numbers of Mo and Sn, and hence correspondingly low contrast expected, nuclei smaller than ca. 10 nm might not be seen in the FESEM. Another, less likely but still possible scenario is that the substrate is covered with a continuous layer, likely to be either amorphous or an adsorbed monolayer. The 1:4.25 Mo:Sn ratio measured could then be roughly estimated to correspond to two monolayers of a Mo–O species. The peaks in Fig. 5 are shifted to lower binding energy by ca. 0.6 eV in comparison to the reported Mo 3d5/2 and 3d3/2 peaks of MoO3 (in Fig. 3a). This indicates a different chemical environment of the Mo in comparison to the normal MoO3 lattice. The same XPS peaks of Mo(V), measured in non-stoichiometric amorphous MoO3 have been measured to be ca. 1 V lower in binding energy compared to the Mo(VI) in stoichiometric films.42
:4.25. Assuming the Mo is in the form of discrete nuclei of a Mo–O material (the most likely expected scenario), this would represent a moderately dense coverage of nuclei with comparable diameter and spacing between the nuclei. Taking into account the relatively close atomic numbers of Mo and Sn, and hence correspondingly low contrast expected, nuclei smaller than ca. 10 nm might not be seen in the FESEM. Another, less likely but still possible scenario is that the substrate is covered with a continuous layer, likely to be either amorphous or an adsorbed monolayer. The 1:4.25 Mo:Sn ratio measured could then be roughly estimated to correspond to two monolayers of a Mo–O species. The peaks in Fig. 5 are shifted to lower binding energy by ca. 0.6 eV in comparison to the reported Mo 3d5/2 and 3d3/2 peaks of MoO3 (in Fig. 3a). This indicates a different chemical environment of the Mo in comparison to the normal MoO3 lattice. The same XPS peaks of Mo(V), measured in non-stoichiometric amorphous MoO3 have been measured to be ca. 1 V lower in binding energy compared to the Mo(VI) in stoichiometric films.42
Fig. 6 shows SEM images of films obtained after a second stage deposition from a solution that contained 0.65 M HNO3 and HMA of varying concentrations (0.1, 0.05, 0.025 and 0.01 M, a–d respectively). With reducing HMA concentration in the solution, an increase in the rod diameter and in the film texture was evident. Increase in HMA concentration results in faster bulk precipitation, and it is probable that the actual concentration of soluble Mo species is higher at lower initial HMA concentration and that the concentration falls due to bulk precipitation much more slowly, thus allowing time for more growth. This is supported by the observed effect of aging of the second stage deposition solution.
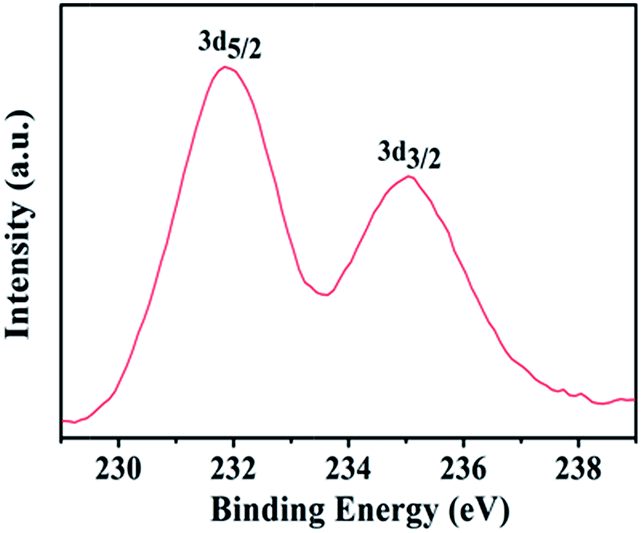 | ||
| Fig. 5 Mo 3d XPS spectrum after the 1st stage of the two step deposition. | ||
The chemical bath deposition process consists of two interlinked steps: nucleation and growth. Often these two processes occur simultaneously, resulting in a large particle size distribution in the film. Separating the nucleation and growth stages leads to a narrower size distribution with better control over the film morphology, as demonstrated previously for ZnO nanorod films.43 Yang et al. showed aligned ZnO nanorod arrays on ZnO nanoparticle-coated Si substrates.44 A pre-nucleated substrate introduces the heterogeneity that assists the growth. The diameter of the rods was primarily determined by the grain size of the ZnO nanoparticles on the seeded substrate. This was also the case where heterogeneous seeding was used, i.e. the seed layer was not ZnO but a hydrated Mn oxide.45
Fig. 6 shows SEM images of films obtained after a second.
 | ||
| Fig. 6 SEM images of the as-deposited films obtained from (a) 0.1 M, (b) 0.065 M (c) 0.025 M and (d) 0.01 M of HMA. | ||
Since homogeneous precipitation gradually occurs in the second stage solution, the reactant concentrations gradually decrease with time. Therefore we expect similar behavior from a fresh low-concentration solution and an aged solution with the same eventual concentration as the fresh low-concentration one. Fig. 7 compares films deposited from a second stage fresh solution (0.025 M HMA and 0.65 M HNO3) and the same solution, aged for 30 min. We found that the second stage deposition happens also when the concentration of both HMA and HNO3 was reduced by 50 times; a continuous increase in rod diameter was obtained with reduction in concentration.
 | ||
| Fig. 7 SEM images of the films obtained from (a) fresh solution and (b) 30 min aged solution for the 2nd stage deposition. The first stage deposition was kept unaltered at 30 min (the inset shows the cross-section micrograph of the same films). | ||
Yet further control of the rod diameter can be obtained by controlling the nucleation at the first stage. Fig. 8 shows films prepared from a first stage deposition solution containing 0.025 M of HMA and 0.65 M HNO3. The substrates were taken out from the deposition solution after 2, 5, 10, 20, 30 and 45 min. At the second stage of the deposition these pre-treated substrates were re-immersed in the same aged solution kept at 85 °C for 30 min. The shorter the time in the first stage bath, the greater is the width of the rods. This can be explained in general by lower nucleation density on the substrate resulting in larger rods since the same amount of available reactant is divided by a smaller number of nuclei. Fig. 9 shows histogram of rod diameters obtained from different films. If nucleation is by a continuous layer of a Mo species, this explanation could still hold assuming that the layer formation occurs slowly.
 | ||
| Fig. 8 SEM image of the films obtained from (a) 2 min, (b) 5 min, (c) 10 min (d) 20 min (e) 30 min and (f) 45 min of 1st stage deposition while the 2nd stage deposition was kept unaltered. | ||
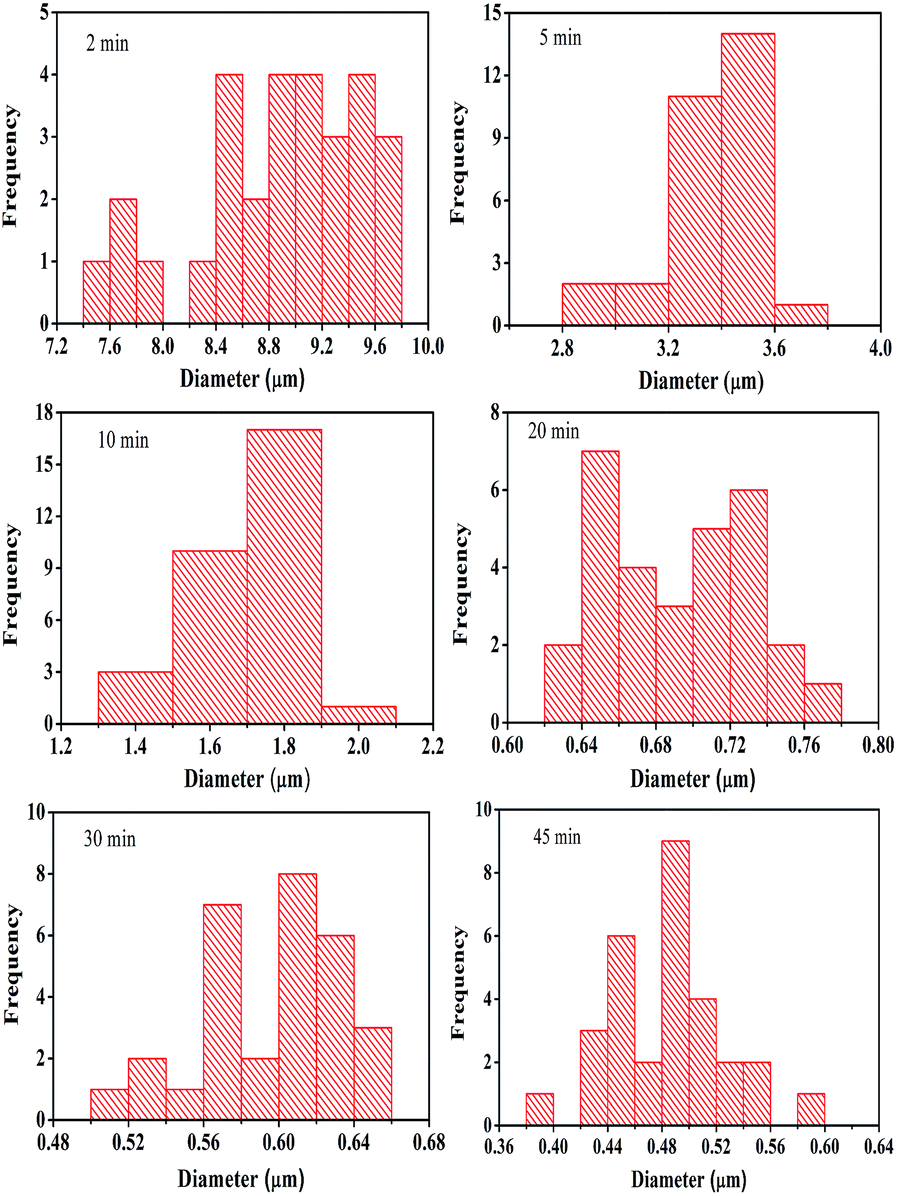 | ||
| Fig. 9 Distribution of nanorod diameters of the films obtained from 2 min, 5 min, 10 min, 20 min, 30 min and 45 min of 1st stage deposition while the 2nd stage deposition was kept unaltered. | ||
We studied the rinsing mechanism in detail to understand the need for the two-stage deposition. Rinsing with water, for example, can cause several changes that might activate the second state deposition process; (a) local change in pH, (b) (another) chemical heterogeneity in the solution, (c) temperature changes at the surface and (d) turbulence.
Rinsing with hot or cold water or even post rinsing heating of the substrate (at 60–70 °C to dry it) has no visible effect in deposition rate. Also stirring the solution or simply shaking it never affected the second stage film growth. The above experiments show that neither temperature changes nor turbulence are likely to initiate the second stage growth.
To understand the effects of local change in pH and the chemical heterogeneity introduced by the rinsing step, we studied the rinsing step extensively by varying the chemical nature of the rinsing medium. There was no difference in film growth or structure if the substrate was rinsed after the first stage process with water (as was normally used) or with acid (both concentrated and dilute), which argues against a pH effect. If the substrate was rinsed with an aqueous solution of high pH (e.g. 1 M NaOH solution), no deposition occurred during the second stage. However, since Mo oxides and oxide salts are soluble in alkaline solutions, this can be explained by dissolution of the Mo–O nuclei formed during the first stage and support the presence of such species after the first stage. However, if the substrate was rinsed with the same solution used in the first stage, there was no deposition in the second stage, regardless of the temperature of the rinsing solution. Therefore, the most likely cause of the rinsing after the first stage leading to deposition during the second stage is some chemical heterogeneity that provides the driving force for deposition in the second stage. Considering the well-known passivation of iron by strong HNO3, a hypothesis is that rinsing the Mo–O nucleation layer removes a passivating film on the Mo–O which prevents continued growth during the first stage. However, since growth does occur after rinsing if the nucleated and rinsed substrate is re-immersed in the original solution, this hypothesis would require some difference between the Mo–O species formed in the first stage and that after rinsing with water that prevents the rinsed Mo–O species from being re-passivated. Regardless of the exact mechanism, it is clear that rinsing at the first stage changes the surface of the nucleation layer sufficiently for deposition to continue to occur.
Photocatalytic degradation of MB dye was observed in presence of h-MoO3 nanorod films. Characteristic absorbance decreased with the time of light exposure as shown in the Fig. 10. We believe that photogenerated charge carriers lead formation of hydroxyl or superoxide radicals that oxidized the MB molecules. The degradation of the MB dye can directly be observed by looking at its color that gets more transparent with time in the presence of h-MoO3 film.
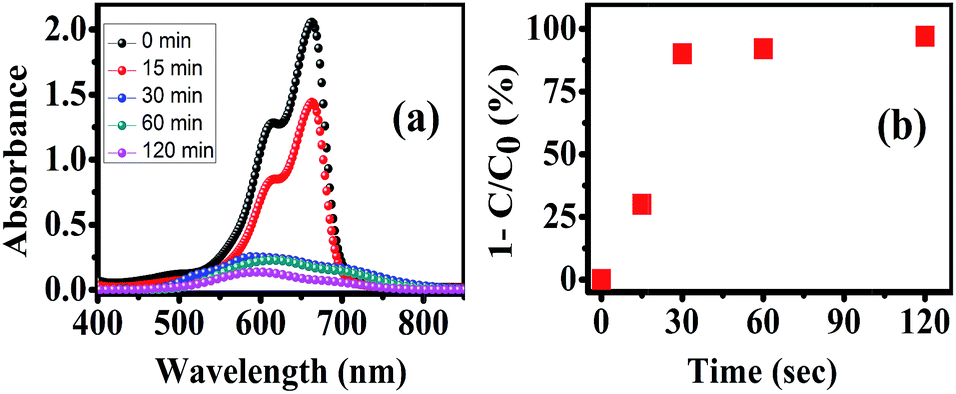 | ||
| Fig. 10 (a) Absorbance plot of different MB solutions after different light exposure time in presence of h-MoO3 nanorod films and (b) plot of degradation percentage of MB with respect to light exposure time. | ||
4. Conclusions
Thin films of highly crystalline hexagonal MoO3 nanorods were deposited by chemical bath deposition. A two-step process where the samples were rinsed between the first and second step was necessary to achieve control over the growth and morphology of the films. Growth at the second stage was believed to be initiated by a chemical change of the nucleation centers formed in the first step caused by the rinsing step. Reasonable photocatalytic degradation of MB dye was observed using h-MoO3 under visible light.Acknowledgements
This work was supported partially by the National Center for Photovoltaics Research and Education (NCPRE) funded by the Ministry of New and Renewable Energy, Government of India.Notes and references
- J. Meyer, M. Kröger, S. Hamwi, F. Gnam, T. Riedl, W. Kowalsky and A. Kahn, Appl. Phys. Lett., 2010, 96, 193302 CrossRef PubMed.
- C.-W. Chu, S.-H. Li, C.-W. Chen, V. Shrotriya and Y. Yang, Appl. Phys. Lett., 2005, 87, 193508 CrossRef PubMed.
- H. Liu, M. Si, Y. Deng, A. T. Neal, Y. Du, S. Najmaei, P. M. Ajayan, J. Lou and P. D. Ye, ACS Nano, 2014, 8, 1031 CrossRef CAS PubMed.
- J. Wang, K. C. Rose and C. M. Lieber, J. Phys. Chem. B, 1999, 103, 8405 CrossRef CAS.
- Y. Bai, X. Liu, L. Chen, Khizar-ul-Haq, M. A. Khan, W. Q. Zhu, X. Y. Jiang and Z. L. Zhang, Microelectron. J., 2007, 38, 1185 CrossRef CAS PubMed.
- D. Qin, J. Liu, Y. Chen, C. Cheng and W. Quan, Phys. Status Solidi A, 2011, 208, 1976 CrossRef CAS.
- M. Wang, Y. Li, H. Huang, E. D. Peterson, W. Nie, W. Zhou, W. Zeng, W. Huang, G. Fang, N. Sun, X. Zhao and D. L. Carroll, Appl. Phys. Lett., 2011, 98, 103305 CrossRef PubMed.
- N. Li, B. E. Lassiter, R. R. Lunt, G. Wei and S. R. Forrest, Appl. Phys. Lett., 2009, 94, 023307 CrossRef PubMed.
- I. Shakir, M. Shahid, H. W. Yang and D. J. Kang, Electrochim. Acta, 2010, 56, 376 CrossRef CAS PubMed.
- R. Liang, H. Cao and D. Qian, Chem. Commun., 2011, 47, 10305 RSC.
- L. Zhou, L. Yang, P. Yuan, J. Zou, Y. Wu and C. Yu, J. Phys. Chem. C, 2010, 114, 21868 CAS.
- P. Meduri, E. Clark, J. H. Kim, E. Dayalan, G. U. Sumanasekera and M. K. Sunkara, Nano Lett., 2012, 12, 1784 CrossRef CAS PubMed.
- S.-H. Lee, Y.-H. Kim, R. Deshpande, P. A. Parilla, E. Whitney, D. T. Gillaspie, K. M. Jones, A. H. Mahan, S. Zhang and A. C. Dillon, Adv. Mater., 2008, 20, 3627 CrossRef CAS.
- D. Y. Kim, J. Subbiah, G. Sarasqueta, F. So, H. Ding, Irfan and Y. Gao, Appl. Phys. Lett., 2009, 95, 093304 CrossRef PubMed.
- C. Girotto, E. Voroshazi, D. Cheyns, P. Heremans and B. P. Rand, ACS Appl. Mater. Interfaces, 2011, 3, 3244 CAS.
- A. Phuruangrat, D. J. Ham, S. Thongtem and J. S. Lee, Electrochem. Commun., 2009, 11, 1740 CrossRef CAS PubMed.
- H. Yang, X. Li, A. Wang, Y. Wang and Y. Chen, Chin. J. Catal., 2014, 35, 140 CrossRef CAS.
- Y. Chen, C. Lu, L. Xu, Y. Ma, W. Hou and J.-J. Zhu, CrystEngComm, 2010, 12, 3740 RSC.
- A. Chithambararaj, N. S. Sanjini, A. C. Bose and S. Velmathi, Catal. Sci. Technol., 2013, 3, 1405 CAS.
- A. M. Taurino, A. Forleo, L. Francioso, P. Siciliano, M. Stalder and R. Nesper, Appl. Phys. Lett., 2006, 88, 152111 CrossRef PubMed.
- T.-Y. Chang, Y.-W. Cheng and P.-T. Lee, Appl. Phys. Lett., 2010, 96, 043309 CrossRef PubMed.
- X. W. Lou and H. C. Zeng, Chem. Mater., 2002, 14, 4781 CrossRef CAS.
- T. M. McEvoy, K. J. Stevenson, J. T. Hupp and X. Dang, Langmuir, 2003, 19, 4316 CrossRef CAS.
- E. M. McCarron, Chem. Commun., 1986, 336 RSC.
- D. Z. Pai, K. K. Ostrikov, S. Kumar, D. A. Lacoste, I. Levchenko and C. O. Laux, Sci. Rep., 2013, 3, 1221 Search PubMed.
- B. Baker, T. P. Feist and E. M. McCarron III, J. Solid State Chem., 1995, 119, 199 CrossRef CAS.
- V. Madhavi, P. Kondaiah, S. S. Rayudu, O. M. Hussain and S. Uthanna, Mater. Express, 2013, 3, 135 CrossRef CAS PubMed.
- J. Zhou, N. S. Xu, S. Z. Deng, J. Chen, J. C. She and Z. L. Wang, Adv. Mater., 2003, 15, 1835 CrossRef CAS.
- I. Navas, R. Vinodkumar, K. J. Lethy, A. P. Detty, V. Ganesan, V. Sathe and V. P. M. Pillai, J. Phys. D: Appl. Phys., 2009, 42, 175305 CrossRef.
- C. Liu, Z. Li and Z. Zhang, Appl. Phys. Lett., 2011, 99, 223104 CrossRef PubMed.
- D. Parviz, M. Kazemeini, A. M. Rashidi and K. Jafari Jozani, J. Nanopart. Res., 2010, 12, 1509 CrossRef CAS.
- J. Song, X. Ni, D. Zhang and H. Zheng, Solid State Sci., 2006, 8, 1164 CrossRef CAS PubMed.
- N. Kumagai, N. Kumagai and K. Tanno, Electrochim. Acta, 1987, 32, 1521 CrossRef CAS.
- H. J. Lunk, H. Hartl, M. A. Hartl, M. J. G. Fait, I. G. Shenderovich, M. Feist, T. A. Frisk, L. L. Daemen, D. Mauder, R. Eckelt and A. A. Gurinov, Inorg. Chem., 2010, 49, 9400 CrossRef CAS PubMed.
- Y. Xu, L. Xie, Y. Zhang and X. Cao, Electron. Mater. Lett., 2013, 9, 693 CrossRef CAS PubMed.
- A. Abdellaoui, L. Martin and A. Donnadieu, Phys. Status Solidi A, 1988, 109, 455 CrossRef CAS.
- S. Deki, A. B. Béléké, Y. Kotani and M. Mizuhata, J. Solid State Chem., 2009, 182, 2362 CrossRef CAS PubMed.
- W. Pan, R. Tian, H. Jin, Y. Guo, L. Zhang, X. Wu, L. Zhang, Z. Han, G. Liu, J. Li, G. Rao, H. Wang and W. Chu, Chem. Mater., 2010, 22, 6202 CrossRef CAS.
- Z. Lei, X. Yang, J. Dong and X. Yi, Chem. Mater., 2009, 21, 5681 CrossRef.
- V. V. Atuchin, T. A. Gavrilova, V. G. Kostrovsky, L. D. Pokrovsky and I. B. Troitskaia, Inorg. Mater., 2008, 44, 622 CrossRef CAS.
- G. R. Patil, R. S. Gaikwad, M. B. Shelar, R. S. Mane, S. H. Hand and B. N. Pawar, Arch. Phys. Res., 2012, 3, 401 CAS.
- M. Rouhani, Y. L. Foo, J. Hobley, J. Pan, G. S. Subramanian, X. Yu, A. Rusydi and S. Gorelik, Appl. Surf. Sci., 2013, 273, 150 CrossRef CAS PubMed.
- H. C. Cheng, C. F. Chen, C. Y. Tsay and J. P. Leu, J. Alloys Compd., 2009, 475, L46 CrossRef CAS PubMed.
- L. L. Yang, Q. X. Zhao, M. Willander and J. H. Yang, J. Cryst. Growth, 2009, 311, 1046 CrossRef CAS PubMed.
- M. Kokotov and G. Hodes, J. Mater. Chem., 2009, 19, 3847 RSC.
| This journal is © The Royal Society of Chemistry 2014 |