
Synthesis and characterization of platinum modified TiO2-embedded carbon nanofibers for solar hydrogen generation†
Abstract
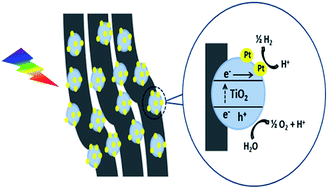
Photocatalytic water splitting is a significant and promising technology to generate hydrogen, an alternative and clean future fuel. The present work describes the preparation and application of a TiO2 based photocatalyst with an effective architecture focused on the key issues of photocatalyst reusability and photocatalytically stable activity. TiO2 nanoparticles were embedded onto carbon nanofibers (CNF) acting as a support. The electrospinning method was adopted for fabrication of TiO2 embedded carbon nanofibers (TiO2/CNF). The photocatalytic activity of the TiO2/CNF under simulated light was improved by Pt co-catalyst photodeposition. The morphological and structural properties of TiO2/CNF and Pt photodeposited TiO2/CNF (Pt–TiO2/CNF) were investigated by field emission scanning electron microscopy (FESEM), high resolution transmission electron microscopy (HRTEM) and powder X-ray diffraction (XRD) techniques. The surface characteristics of these materials were investigated by X-ray photoelectron spectroscopy (XPS), photocurrent and photoluminescence (PL) measurements were also used to elaborate the effect of Pt photodeposition. The reusability and photocatalytic stability of Pt–TiO2/CNF was tested by the generation of approximately the same amounts of photocatalytic hydrogen with the same sample under the same conditions of illumination. The highest production rate of photocatalytic hydrogen generation achieved under simulated light was 3.5 μmol (after 3 hours of illumination with 0.02 g of photocatalyst). These tests show that Pt–TiO2/CNF is a reusable photocatalyt with a promising stable photocatalytic performance for solar energy conversion applications.
 Please wait while we load your content...
Please wait while we load your content...

