
Investigation of hydrogen bonding patterns in a series of multi-component molecular solids formed by tetrabromoterephthalic acid with selected N-heterocycles†
Lei Wang*,
Yanjing Hu,
Wenyan Xu,
Yanyan Pang,
Faqian Liu and
Yu Yang*
Key Laboratory of Eco-chemical Engineering, Ministry of Education, Laboratory of Inorganic Synthesis and Applied Chemistry, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, P. R. China. E-mail: inorchemwl@126.com; yangyu9039@163.com; Fax: +86-532-840-22681
First published on 16th September 2014
Abstract
The supramolecular reactions of tetrabromoterephthalic acid (H2-TBTA) with a series of N-heterocycles afforded eight new complexes, namely, [(H2-BTAH)2·(TBTA)·(H2-TBTA)] (1), [(H2-Bim)2·(TBTA)·(H2-TBTA)·2H2O] (2), [(H-8-HQ)2·(TBTA)·3H2O] (3), [(5-NO2-phen)2·(H2-TBTA)] (4), [(4,6-DHP)2·(H2-TBTA)·2H2O] (5), [(H2-2,4-DMI)2·(TBTA)·(H2-TBTA)2] (6), [(H2-3,5-DMP)2·(TBTA)] (7), and [(H-4-CNpy)2·(TBTA)·(H2-TBTA)] (8) (H-BTAH = 1H-benzotriazole, H-Bim = 1H-benzimidazole, 8-HQ = 8-hydroxyquinoline, 5-NO2-phen = 5-nitro-1,10-phenanthroline, 4,6-DHP = 4,6-dihydroxypyrimidine, H-2,4-DMI = 2,4-dimethylimidazole, H-3,5-DMP = 3,5-dimethylpyrazole, and 4-CNpy = 4-cyanopyridine), which have been prepared under mild and identical reaction conditions in a mixture of distilled water and ethanol. All the complexes were fully characterized using single crystal X-ray diffraction analysis, elemental analysis, infrared spectroscopy (IR), and thermogravimetric analysis (TGA). Combining the various N-containing ligands and the diversity of the hydrogen bonds, the eight crystals display amusing structural characteristics. Among these complexes, complex 3 forms a three-dimensional (3D) network through the C–H⋯Br bonds, while the O–H⋯Br bonds facilitate the 3D construction of compound 2. Complexes 4–8 generate 3D supramolecular structures by utilizing a large number of hydrogen bonds. In crystal 1, the π–π stacking interactions play an important part in the 3D network. The thermal stability of crystals 1–8 has been investigated by thermogravimetric analysis (TGA) of mass loss.
Introduction
The rational design and synthesis of organic solids from molecular components is one of the main areas of focus in the field of crystal engineering,1 not only because of their intriguing structures but also because of their potential applications in drug delivery,2 sensing,3 and nonlinear optics.4 Self-assembly is the fundamental feature of natural and biological processes with noncovalent interactions, such as hydrogen bonds, electrostatic interactions, van der Waals forces, π–π stacking interactions, and halogen bonds,6 acting as tools to accomplish these tasks.5 Currently, strong hydrogen bonds such as O–H⋯O and N–H⋯O are very prevalent in controlling molecular assembly during crystallization.7 Furthermore, weak C–H⋯O and C–H⋯N hydrogen bonds also play an important part in distorting and modifying the predicted structure.8 In the last two decades, the halogen bonds, especially the C–H⋯halogen hydrogen bonds and halogen⋯halogen interactions, have gained widespread attention,9 although the first unequivocal report on halogen bonding can be traced back to 1863 when Guthrie described the formation of the complex NH3⋯I2.10Nowadays, more and more researchers are focusing on halogen-substituted compounds,11 because chemists have recognized that the halogen bonds play a significant role in crystal packing in many halogen-containing organic compounds12 due to their highly directional, as well as fully hydrophobic characteristics.13 Tetrahalogen-substituted dicarboxylic acids are good ligands and have a lot of advantages, which are applied to metal-organic frameworks (MOFs) and supramolecular chemistry.14 However, to date, the systematic studies concerned with tetrahalogen-substituted dicarboxylic acids are still relatively scarce. The results of our previous studies14k,l show that tetrafluoroterephthalic acid exhibits the special ability to form organic solids through noncovalent interactions, particularly the halogen bond C–H⋯F.
In order to continue our previous work and explore the role of the halogen bonds in the synthesis of the supramolecular, we selected tetrabromoterephthalic acid (H2-TBTA) as the dicarboxylic acid ligand, and 1H-benzotriazole, 1H-benzimidazole, 8-hydroxyquinoline, 5-nitro-1,10-phenanthroline, 4,6-dihydroxypyrimidine, 2,4-dimethylimidazole, 3,5-dimethylpyrazole and 4-cyanopyridine, which are employed as N-heterocycles (Scheme 1). In this study, eight new complexes have been obtained through noncovalent interactions. Moreover, the structural characterization and the thermal stabilities of these complexes have been investigated in the solid state.
 | ||
| Scheme 1 Molecular structures of compounds used in this work. | ||
Experimental section
Materials
All the reagents and solvents were commercially available and used as received without further purification. 1H-Benzotriazole, 1H-benzimidazole, 8-hydroxyquinoline, 5-nitro-1,10-phenanthroline, 4,6-dihydroxypyrimidine, 2,4-dimethylimidazole, 3,5-dimethylpyrazole, 4-cyanopyridine and tetrabromoterephthalic acid were obtained from Energy Chemical.Physical measurements
Melting point measurements were carried out using a WRS-1B digital thermal apparatus without correction, and they refer to the temperature at the start of the melt. The FT-IR spectra were recorded on a Nicolet Impact 410 FTIR in the range of 4000–400 cm−1 using the KBr disc technique. Absorptions are denoted as follows: strong (s), medium (m), and weak (w) in the synthesis section. Carbon, hydrogen, and nitrogen contents were determined with a Perkin-Elmer 2400 elemental analyzer. Thermogravimetric analysis (TGA) was performed from room temperature to 900 °C using a Perkin-Elmer TGA-7 TG analyzer with a heating rate of 10 °C min−1 in a N2 atmosphere. Eight novel crystals were obtained as follows.Synthesis of the complexes 1–8
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), and dried under vacuum. Crystals were obtained in 58.7% yield by filtration and found to be suitable for single-crystal X-ray diffraction analysis. m.p.: 204 °C. Anal. calcd (%) for C21H21Br4N2O7 (1): C 34.38, H 2.86, N 3.82; found: C 34.16, H 3.04, N 3.66. IR (KBr pellet, cm−1): 3436(m), 3111(w), 2963(w), 2918(w), 2852(w), 2520(w), 1616(m), 1405(m), 1328(m), 1304(m), 1237(m), 1221(m), 1180(m), 1136(m), 1085(m), 1008(w), 996(w), 937(w), 876(m), 805(m), 749(s), 742(s), 661(m), 621(s), 563(s), 536(s), 520(s), 425(m).:1), and then dried in vacuum desiccators. Crystals were found to be suitable for single-crystal X-ray diffraction analysis. m.p.: 246 °C. Anal. calcd (%) for C30H20Br8N4O10 (2): C 29.13, H 1.62, N 4.53; found: C 28.95, H 1.68, N 4.48. IR (KBr pellet, cm−1): 3586(m), 3385(m), 3154(w), 3075(w), 2970(w), 2829(m), 2755(m), 2684(m), 2593(m), 1711(m), 1635(s), 1626(s), 1611(s), 1449(m), 1422(m), 1378(m), 1330(s), 1303(s), 1241(s), 1191(m), 1153(w), 1081(m), 1007(w), 985(w), 923(w), 876(w), 848(m), 836(m), 785(w), 746(s), 691(w), 619(s), 608(m), 576(m), 557(m), 523(m), 420(m).:1) and dried under vacuum. Crystals obtained were suitable for single-crystal X-ray diffraction analysis. m.p.: 266 °C. Anal. calcd (%) for C26H22Br4N2O9 (3): C 37.77, H 2.66, N 3.39; found: C 37.14, H 2.88, N 3.12. IR (KBr pellet, cm−1): 3450(m), 3236(m), 3070(m), 3056(m), 3023(m), 2918(m), 2792(m), 2690(m), 2583(m), 1636(m), 1600(s), 1585(s), 1564(s), 1504(m), 1475(m), 1423(s), 1400(s), 1323(s), 1301(s), 1264(m), 1212(m), 1177(m), 1143(m), 1099(m), 1083(m), 1035(w), 987(m), 938(w), 890(w), 824(s), 802(m), 773(m), 713(m), 701(m), 623(m), 564(s), 542(m), 520(m), 489(m), 475(m), 416(m).:1 molar ratio and dissolved in ethanol–distilled water solution (v/v = 1:1, 10 mL), the solution was stirred for 15 min and a turbid liquid was obtained, which was maintained overnight at room temperature. The next day, the turbid liquid was filtered and the clear homogeneous solution was retained at room temperature for slow evaporation. Good quality orange crystals, which are suitable for diffraction, were obtained after one week as the solution slowly evaporated at room temperature. The crystals were filtered from the mother liquor, washed with ethanol–distilled water solution (v/v = 1:1), and dried in vacuum desiccators. Crystals were obtained in 56.7% yield and were found to be suitable for single-crystal X-ray diffraction analysis. m.p.: 236 °C. Anal. calcd (%) for C32H16Br4N6O8 (4): C 41.20, H 1.72, N 9.01; found: C 40.79, H 1.89, N 8.87. IR (KBr pellet, cm−1): 3436(m), 3217(w), 3096(w), 2872(w), 2570(w), 2477(w), 1713(s), 1637(s), 1617(s), 1593(m), 1541(s), 1531(s), 1497(m), 1467(m), 1450(w), 1435(w), 1417(w), 1346(m), 1332(s), 1306(s), 1231(s), 1177(s), 1086(s), 979(m), 920(m), 875(m), 830(m), 817(m), 794(s), 777(s), 723(m), 701(m), 620(m), 557(s), 525(m), 486(m), 413(m).:1), and dried in vacuum desiccators. Note that the crystals were found to be suitable for single-crystal X-ray diffraction analysis. m.p.: 214 °C. Anal. calcd (%) for C16H14Br4N4O10 (5): C 25.88, H 1.89, N 7.55; found: C 25.76, H 2.04, N 7.36. IR (KBr pellet, cm−1): 3532(m), 3381(m), 3078(m), 2727(m), 2611(m), 1997(w), 1683(s), 1660(s), 1592(s), 1568(m), 1457(w), 1376(m), 1358(m), 1334(m), 1307(s), 1264(m), 1207(m), 1138(m), 1088(m), 992(w), 925(m), 818(m), 788(m), 733(w), 695(m), 600(m), 574(m), 527(s), 459(s), 445(m).:1), and dried under vacuum. m.p.: 222 °C. Anal. calcd (%) for C34H22Br12N4O12 (6): C 24.92, H 1.34, N 3.42; found: C 24.76, H 1.50, N 3.28. IR (KBr pellet, cm−1): 3436(m), 3150(m), 3097(m), 2975(m), 2931(m), 2755(m), 2667(w), 2593(w), 1749(s), 1690(m), 1649(m), 1559(m), 1437(m), 1391(s), 1333(s), 1306(s), 1241(s), 1089(m), 1079(m), 1008(m), 968(m), 885(m), 798(m), 787(m), 719(m), 692(m), 628(m), 589(m), 553(m), 520(m).:1, 10 mL) containing 3,5-dimethylpyrazole (19.2 mg, 0.20 mmol) was added H2-TBTA (42.8 mg, 0.10 mmol) with constant stirring for 15 min. The clear and homogeneous solution was slowly evaporated at room temperature, and block colorless crystals were obtained two weeks later. The crystals were filtered from the mother liquor and washed with acetone–distilled water solution (v/v = 1:1), and dried under vacuum. Crystals were obtained in 58% yield by filtration and were found to be suitable for single-crystal X-ray diffraction analysis. m.p.: 260 °C. Anal. calcd (%) for C18H18Br4N4O4 (7): C 32.05, H 2.67, N 8.31; found: C 31.84, H 2.88, N 8.20. IR (KBr pellet, cm−1): 3458(m), 3430(m), 3139(w), 2982(m), 2917(m), 2680(m), 1723(m), 1613(s), 1464(m), 1422(s), 1362(m), 1331(s), 1306(s), 1232(s), 1160(m), 1083(s), 1016(m), 980(m), 890(m), 849(m), 816(m), 786(m), 727(m), 695(m), 578(s), 560(s), 523(m).:1), and dried under vacuum. Crystals were obtained in 58% yield by filtration and were found to be suitable for single-crystal X-ray diffraction analysis. m.p.: 276 °C. Anal. calcd (%) for C28H12Br8N4O8 (8): C 28.68, H 1.02, N 4.78; found: C 28.54, H 1.48, N 4.56. IR (KBr pellet, cm−1): 3436(m), 3109(w), 3060(w), 2490(w), 1949(w), 1729(m), 1634(s), 1497(m), 1381(m), 1333(m), 1308(s), 1278(s), 1254(s), 1212(s), 1085(m), 1077(m), 958(m), 835(s), 774(m), 753(m), 734(m), 721(w), 657(w), 577(s), 555(m), 523(m), 467(s).
:1), and dried under vacuum. Crystals were obtained in 58.7% yield by filtration and found to be suitable for single-crystal X-ray diffraction analysis. m.p.: 204 °C. Anal. calcd (%) for C21H21Br4N2O7 (1): C 34.38, H 2.86, N 3.82; found: C 34.16, H 3.04, N 3.66. IR (KBr pellet, cm−1): 3436(m), 3111(w), 2963(w), 2918(w), 2852(w), 2520(w), 1616(m), 1405(m), 1328(m), 1304(m), 1237(m), 1221(m), 1180(m), 1136(m), 1085(m), 1008(w), 996(w), 937(w), 876(m), 805(m), 749(s), 742(s), 661(m), 621(s), 563(s), 536(s), 520(s), 425(m).:1), and then dried in vacuum desiccators. Crystals were found to be suitable for single-crystal X-ray diffraction analysis. m.p.: 246 °C. Anal. calcd (%) for C30H20Br8N4O10 (2): C 29.13, H 1.62, N 4.53; found: C 28.95, H 1.68, N 4.48. IR (KBr pellet, cm−1): 3586(m), 3385(m), 3154(w), 3075(w), 2970(w), 2829(m), 2755(m), 2684(m), 2593(m), 1711(m), 1635(s), 1626(s), 1611(s), 1449(m), 1422(m), 1378(m), 1330(s), 1303(s), 1241(s), 1191(m), 1153(w), 1081(m), 1007(w), 985(w), 923(w), 876(w), 848(m), 836(m), 785(w), 746(s), 691(w), 619(s), 608(m), 576(m), 557(m), 523(m), 420(m).:1) and dried under vacuum. Crystals obtained were suitable for single-crystal X-ray diffraction analysis. m.p.: 266 °C. Anal. calcd (%) for C26H22Br4N2O9 (3): C 37.77, H 2.66, N 3.39; found: C 37.14, H 2.88, N 3.12. IR (KBr pellet, cm−1): 3450(m), 3236(m), 3070(m), 3056(m), 3023(m), 2918(m), 2792(m), 2690(m), 2583(m), 1636(m), 1600(s), 1585(s), 1564(s), 1504(m), 1475(m), 1423(s), 1400(s), 1323(s), 1301(s), 1264(m), 1212(m), 1177(m), 1143(m), 1099(m), 1083(m), 1035(w), 987(m), 938(w), 890(w), 824(s), 802(m), 773(m), 713(m), 701(m), 623(m), 564(s), 542(m), 520(m), 489(m), 475(m), 416(m).:1 molar ratio and dissolved in ethanol–distilled water solution (v/v = 1:1, 10 mL), the solution was stirred for 15 min and a turbid liquid was obtained, which was maintained overnight at room temperature. The next day, the turbid liquid was filtered and the clear homogeneous solution was retained at room temperature for slow evaporation. Good quality orange crystals, which are suitable for diffraction, were obtained after one week as the solution slowly evaporated at room temperature. The crystals were filtered from the mother liquor, washed with ethanol–distilled water solution (v/v = 1:1), and dried in vacuum desiccators. Crystals were obtained in 56.7% yield and were found to be suitable for single-crystal X-ray diffraction analysis. m.p.: 236 °C. Anal. calcd (%) for C32H16Br4N6O8 (4): C 41.20, H 1.72, N 9.01; found: C 40.79, H 1.89, N 8.87. IR (KBr pellet, cm−1): 3436(m), 3217(w), 3096(w), 2872(w), 2570(w), 2477(w), 1713(s), 1637(s), 1617(s), 1593(m), 1541(s), 1531(s), 1497(m), 1467(m), 1450(w), 1435(w), 1417(w), 1346(m), 1332(s), 1306(s), 1231(s), 1177(s), 1086(s), 979(m), 920(m), 875(m), 830(m), 817(m), 794(s), 777(s), 723(m), 701(m), 620(m), 557(s), 525(m), 486(m), 413(m).:1), and dried in vacuum desiccators. Note that the crystals were found to be suitable for single-crystal X-ray diffraction analysis. m.p.: 214 °C. Anal. calcd (%) for C16H14Br4N4O10 (5): C 25.88, H 1.89, N 7.55; found: C 25.76, H 2.04, N 7.36. IR (KBr pellet, cm−1): 3532(m), 3381(m), 3078(m), 2727(m), 2611(m), 1997(w), 1683(s), 1660(s), 1592(s), 1568(m), 1457(w), 1376(m), 1358(m), 1334(m), 1307(s), 1264(m), 1207(m), 1138(m), 1088(m), 992(w), 925(m), 818(m), 788(m), 733(w), 695(m), 600(m), 574(m), 527(s), 459(s), 445(m).:1), and dried under vacuum. m.p.: 222 °C. Anal. calcd (%) for C34H22Br12N4O12 (6): C 24.92, H 1.34, N 3.42; found: C 24.76, H 1.50, N 3.28. IR (KBr pellet, cm−1): 3436(m), 3150(m), 3097(m), 2975(m), 2931(m), 2755(m), 2667(w), 2593(w), 1749(s), 1690(m), 1649(m), 1559(m), 1437(m), 1391(s), 1333(s), 1306(s), 1241(s), 1089(m), 1079(m), 1008(m), 968(m), 885(m), 798(m), 787(m), 719(m), 692(m), 628(m), 589(m), 553(m), 520(m).:1, 10 mL) containing 3,5-dimethylpyrazole (19.2 mg, 0.20 mmol) was added H2-TBTA (42.8 mg, 0.10 mmol) with constant stirring for 15 min. The clear and homogeneous solution was slowly evaporated at room temperature, and block colorless crystals were obtained two weeks later. The crystals were filtered from the mother liquor and washed with acetone–distilled water solution (v/v = 1:1), and dried under vacuum. Crystals were obtained in 58% yield by filtration and were found to be suitable for single-crystal X-ray diffraction analysis. m.p.: 260 °C. Anal. calcd (%) for C18H18Br4N4O4 (7): C 32.05, H 2.67, N 8.31; found: C 31.84, H 2.88, N 8.20. IR (KBr pellet, cm−1): 3458(m), 3430(m), 3139(w), 2982(m), 2917(m), 2680(m), 1723(m), 1613(s), 1464(m), 1422(s), 1362(m), 1331(s), 1306(s), 1232(s), 1160(m), 1083(s), 1016(m), 980(m), 890(m), 849(m), 816(m), 786(m), 727(m), 695(m), 578(s), 560(s), 523(m).:1), and dried under vacuum. Crystals were obtained in 58% yield by filtration and were found to be suitable for single-crystal X-ray diffraction analysis. m.p.: 276 °C. Anal. calcd (%) for C28H12Br8N4O8 (8): C 28.68, H 1.02, N 4.78; found: C 28.54, H 1.48, N 4.56. IR (KBr pellet, cm−1): 3436(m), 3109(w), 3060(w), 2490(w), 1949(w), 1729(m), 1634(s), 1497(m), 1381(m), 1333(m), 1308(s), 1278(s), 1254(s), 1212(s), 1085(m), 1077(m), 958(m), 835(s), 774(m), 753(m), 734(m), 721(w), 657(w), 577(s), 555(m), 523(m), 467(s).X-ray data collection and structure determinations
Crystallographic diffraction data of complexes 1–8 were obtained on an Agilent Technologies Gemini A Ultra Atlas CCD with graphite-monochromatized Mo-Kα radiation (λ = 0.71073 Å) at 293 K. Absorption corrections were applied using multi-scan technique. There was no evidence of crystal decay during the data collection for all complexes. All the structures were solved by Direct Method of SHELXS-97 and refined by full-matrix least-squares techniques based on F2 with the SHELXL-97 (ref. 15) crystallographic software package. The hydrogen atoms were placed at calculated positions and refined as riding atoms with isotropic displacement parameters.†Results and discussion
Preparation of compounds 1–8
In our initial crystallizations, we varied the stoichiometries of tetrabromoterephthalic acid and base-type reagents (1:2, 1:1, and 2:1) in parallel solution experiments. However, for these three different ratios, we obtained eight novel crystals.
Reactions of tetrabromoterephthalic acid with 1H-benzotriazole (1:2), 1H-benzimidazole (1:1), 8-hydroxyquinoline (1:2), 5-nitro-1,10-phenanthroline (2:1), 4,6-dihydroxypyrimidine (2:1), 2,4-dimethylimidazole (1:1), 3,5-dimethylpyrazole (1:2), and 4-cyanopyridine (2:1) results in crystals. The crystals structures of all eight materials (1–8) were obtained in different ratios with the same solvent. Eight new solids were obtained from different solvent combinations: two cocrystals with 5-nitro-1,10-phenanthroline and 4,6-dihydroxypyrimidine (5); two hydrous salts with 1H-benzimidazole (2), 8-hydroxyquinoline (3); four salts with 1H-benzotriazole (1), 2,4-dimethylimidazole (6), 3,5-dimethylpyrazole (7), and 4-cyanopyridine (8). Moreover, they exhibit many common features, especially in the formation of halogen bonds. Similar to most of the supramoleculars previously reported these crystals contain a large number of hydrogen bond networks in which the tetrabromoterephthalic acid and base components form a series of possible synthons. The schematic representations of different kinds of hydrogen bonding synthons I–XIII related to this work are summarized in Scheme 2. More synthons XIV–XXXI related to this work are displayed in the ESI.† The crystallographic parameters are summarized in Table 1, whereas hydrogen bond geometries of 1–8 are listed in Table 2. Now, we discuss the structural aspects of these new multi-component crystals.
 | ||
| Scheme 2 Supramolecular synthons. | ||
| Compound | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| a R1 = ΣΔFoΔ − ΔFcΔ/ΣΔFoΔ.b wR2 = [Σw(Fo2 − Fc2)2/Σw(Fo2)2]1/2. | ||||||||
| Empirical formula | C28H14Br8N6O8 | C30H20Br8N4O10 | C26H22Br4N2O9 | C32H16Br4N6O8 | C16H14Br4N4O10 | C34H22Br12N4O12 | C18H18Br4N4O4 | C28H12Br8N4O8 |
| M | 1201.65 | 1235.70 | 826.09 | 932.11 | 741.91 | 1637.36 | 673.96 | 1171.62 |
| Crystal system | Monoclinic | Triclinic | Triclinic | Monoclinic | Monoclinic | Triclinic | Triclinic | Triclinic |
| Space group | P21/c | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/c | P21/n | P |
P |
P |
| a/Å | 11.5626(5) | 8.9459(10) | 10.2368(6) | 17.4367(5) | 7.8320(4) | 9.0669(4) | 8.1803(8) | 8.8990(5) |
| b/Å | 13.4148(4) | 9.0271(9) | 11.7277(7) | 11.5949(2) | 16.5732(7) | 11.1569(4) | 8.7685(7) | 9.0236(7) |
| c/Å | 11.3223(5) | 12.5435(11) | 12.9747(7) | 18.1466(11) | 8.9252(4) | 12.8684(4) | 8.9983(10) | 12.0472(8) |
| α/° | 90 | 72.408(9) | 72.509(5) | 90 | 90 | 106.546(3) | 115.443(10) | 105.180(7) |
| β/° | 100.498(5) | 87.358(9) | 86.189(5) | 121.166(3) | 100.132(4) | 106.524(4) | 102.074(9) | 109.306(6) |
| γ/° | 90 | 73.572(10) | 73.802(5) | 90 | 90 | 100.289(4) | 91.498(8) | 96.573(6) |
| V/Å3 | 1726.81(12) | 925.31(18) | 1426.41(15) | 3139.3(2) | 1140.44(9) | 1147.58(9) | 565.01(11) | 859.41(12) |
| Z | 2 | 1 | 2 | 4 | 2 | 1 | 1 | 1 |
| T/K | 293 | 293 | 293 | 293 | 293 | 293 | 293 | 293 |
| Dc/g cm−3 | 2.311 | 2.218 | 1.923 | 1.972 | 2.161 | 2.369 | 1.981 | 2.264 |
| μ/mm−1 | 9.348 | 8.728 | 5.699 | 5.193 | 7.119 | 10.533 | 7.155 | 9.386 |
| F(000) | 1136.0 | 588.0 | 808.0 | 1816.0 | 716.0 | 770.0 | 326.0 | 552.0 |
| h, k, lmax | 13, 15, 13 | 10, 10, 14 | 14, 16, 18 | 20, 13, 21 | 10, 23, 12 | 10, 13, 15 | 9, 10, 10 | 12, 12, 16 |
| R1a | 0.0352 | 0.0366 | 0.0400 | 0.0382 | 0.0388 | 0.0322 | 0.0308 | 0.0612 |
| wR2b (all data) | 0.0708 | 0.0829 | 0.0808 | 0.0931 | 0.0813 | 0.0610 | 0.0729 | 0.1661 |
| S (GOF on F2) | 1.020 | 1.041 | 0.999 | 1.036 | 1.064 | 1.018 | 1.082 | 1.033 |
| D-H⋯A (Å) (symmetry code) | D–H (Å) | H⋯A (Å) | D⋯A (Å) | D–H⋯A (deg) |
|---|---|---|---|---|
| 1O1–H1A⋯O4 (1 + x, y, −1 + z) | 0.989 | 1.636 | 2.618 | 171.3 |
| N3–H3⋯O4 (2 − x, 1 − y, −z) | 0.815 | 2.012 | 2.775 | 155.4 |
| N1–H1⋯O3 (1 + x, y, −1 + z) | 1.021 | 1.594 | 2.614 | 176.3 |
| 2O4–H4⋯O2 (x, 1 + y, z) | 0.820 | 1.752 | 2.566 | 171.3 |
| O5–H5B⋯O1 (x, 1 + y, z) | 0.850 | 1.949 | 2.785 | 167.2 |
| N2–H2⋯O5 (x, 1 + y, z) | 0.860 | 1.885 | 2.731 | 167.2 |
| N1–H1⋯O2 (x, 1 + y, 1 + z) | 0.860 | 1.864 | 2.696 | 162.3 |
| O5–H5A⋯Br1 (x, 1 + y, z) | 0.850 | 3.146 | 3.559 | 112.5 |
| 3O9–H9A⋯O2 (x, y, z) | 0.850 | 1.964 | 2.812 | 175.1 |
| O9–H9B⋯O3 (x, y, 1 + z) | 0.850 | 2.026 | 2.872 | 173.2 |
| O8–H8B⋯O9 (x, −1 + y, z) | 0.787 | 1.966 | 2.732 | 164.2 |
| O8–H8A⋯O7 (1 − x, 1 − y, −z) | 0.747 | 2.011 | 2.754 | 173.2 |
| O6–H6⋯O1 (x, y, z) | 0.820 | 1.689 | 2.508 | 177.3 |
| C37–H37⋯Br1 (x, y, 1 + z) | 0.930 | 3.110 | 3.769 | 129.4 |
| 4O4–H4⋯N4 (−x, −y, −z) | 0.814 | 1.863 | 2.674 | 174.2 |
| C50–H50⋯O7 (x, y, z) | 0.930 | 2.812 | 3.440 | 125.9 |
| C46–H46⋯O3 (x, y, 1 + z) | 0.930 | 2.877 | 3.753 | 157.5 |
| 5N1–H1⋯O3 (−x, −y, −1 − z) | 0.860 | 1.870 | 2.716 | 167.3 |
| O3–H3A⋯O5 (1 − x, 1 − y, −z) | 0.850 | 1.944 | 2.754 | 159.0 |
| N2–H2⋯O5 (1 − x, 1 − y, −z) | 0.860 | 1.862 | 2.717 | 172.7 |
| O2–H2A⋯O4 (x, 2 + y, z) | 0.820 | 1.727 | 2.523 | 163.3 |
| 6O6–H6⋯O2 (1 + x, y, z) | 0.820 | 1.806 | 2.621 | 172.4 |
| O3–H3⋯O1 (x, 2 + y, z) | 0.820 | 1.672 | 2.491 | 177.6 |
| N1–H1⋯O2 (x, 2 + y, z) | 0.860 | 1.981 | 2.827 | 167.8 |
| N2–H2⋯O4 (1 + x, y, z) | 0.860 | 1.991 | 2.814 | 159.9 |
| 7N2–H2⋯O1 (1 + x, y, z) | 0.860 | 1.824 | 2.655 | 161.8 |
| C14–H14B⋯O2 (1 + x, y, z) | 0.960 | 2.488 | 3.384 | 155.4 |
| C19–H19A⋯O2 (1 + x, y, z) | 0.960 | 2.839 | 3.384 | 116.9 |
| C19–H19C⋯O2 (1 + x, y, z) | 0.960 | 3.324 | 3.729 | 107.6 |
| 8C30–H30⋯O4 (x, y, 1 + z) | 0.930 | 2.690 | 3.221 | 117.0 |
| C32–H32⋯N2 (−x, −y, −z) | 0.930 | 2.470 | 3.361 | 160.5 |
| O4–H4⋯O1 (x, y, z) | 0.820 | 1.755 | 2.537 | 158.7 |
| N1–H1⋯O1 (1 + x, y, z) | 1.159 | 1.446 | 2.571 | 161.2 |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond distances. The lengths of two C–O bonds for the tetrabromoterephthalic acid molecule are obviously different (1.300 Å for C–OH and 1.211 Å for CO), whereas the two C–O bonds for tetrabromoterephthalic acid dianion are very similar (1.254 Å and 1.252 Å), showing that one acid is neutral and the other one is fully deprotonated. The exocyclic bond lengths C20–C24 and C17–C22 are elongated to 1.506 Å and 1.518 Å, longer than those of cyclic C–C bonds (average 1.389 Å). The bond lengths C–Br are on average 1.883 Å. Note that within the 1H-benzotriazole subunit, the triazole ring deviates by 1.523° from the benzene ring; within the acid molecule and acid dianion components, the dihedral angle between two carboxyl planes is 6.966°; and within the tetrabromoterephthalic acid subunits, the carboxyl plane and the benzene ring deviate by 80.529° and 81.103°, respectively.
O bond distances. The lengths of two C–O bonds for the tetrabromoterephthalic acid molecule are obviously different (1.300 Å for C–OH and 1.211 Å for CO), whereas the two C–O bonds for tetrabromoterephthalic acid dianion are very similar (1.254 Å and 1.252 Å), showing that one acid is neutral and the other one is fully deprotonated. The exocyclic bond lengths C20–C24 and C17–C22 are elongated to 1.506 Å and 1.518 Å, longer than those of cyclic C–C bonds (average 1.389 Å). The bond lengths C–Br are on average 1.883 Å. Note that within the 1H-benzotriazole subunit, the triazole ring deviates by 1.523° from the benzene ring; within the acid molecule and acid dianion components, the dihedral angle between two carboxyl planes is 6.966°; and within the tetrabromoterephthalic acid subunits, the carboxyl plane and the benzene ring deviate by 80.529° and 81.103°, respectively.
 | ||
| Fig. 1 (a) Molecular structure of 1 with atom labeling of the asymmetric unit; (b) the infinite 1D hydrogen bonding chain via O–H⋯O hydrogen bonds (the hydrogen bonds are indicated as broken lines in this and the subsequent figures); (c) perspective view of the 2D hydrogen-bonded layer; (d) the resultant 2D layer in two colours; (e) 3D network via π–π stacking interactions; (f) view of the π–π stacking interactions of H2-TBTA molecular and 1H-BT belong to different layer (O, red; N, blue; C, gray; H, turquoise; Br, violet in this and the subsequent figures). | ||
Complex 1 exhibits a one-dimensional chain in Fig. 1b. These acid molecules are linked through the O1–H1A⋯O4 hydrogen bonds, and form oxygen–hydrogen–oxygen bonds that are balanced perfectly in a V-shape at a 110.928(2)° degree angle. Furthermore, the adjacent 1D chains are assembled into 2D layers in the ab-plane via N–H⋯O interactions between acid and base molecules, which is shown in the Fig. 1c. In order to display this more clearly, we show that the 1D chains in pink and the 1H-benzotriazole molecules in bright green, as shown in Fig. 1d. Significantly, the adjacent benzene rings of the 2D layers are almost parallel (the dihedral angle between them is 3.450°), and the distance between the central point of the benzene rings is 3.6439(2) Å. It is within the range of π–π stacking interactions (3.3–3.8 Å); hence, we conclude that π–π stacking interactions exist. Further analysis of the crystal packing indicated that the π–π stacking interactions link two adjoining sheets to provide a 3D network structure (Fig. 1e). Fig. 1f provides the detailed analysis of the π–π stacking interactions of the 3D network. Note that synthons I R44(16) and XIV R88(52) are formed properly and are shown in Scheme 2.
space group. The asymmetric unit of 2 contains one H2-Bim+ cation, half a molecule of H2-TBTA, half of a TBTA2− dianion and one H2O molecule (see Table 1). Within 2, similar to compound 1, the tetrabromoterephthalic acid molecule is fully deprotonated with one hydroxyl proton transferred to the H-Bim molecule. The state of the carboxylic moiety (neutral or ionic) can be found through the C–O and CO bond distances. The lengths of two C–O bonds for tetrabromoterephthalic acid molecule are obviously different (1.304 Å for C–OH and 1.205 Å for CO), while the two C–O bonds for tetrabromoterephthalic acid dianion are very similar (1.262 Å and 1.228 Å), showing that one acid is neutral and the other is fully deprotonated. The exocyclic bond lengths C4–C19 and C8–C11 are elongated to 1.518 Å and 1.525 Å, longer than those of cyclic C–C bonds (average 1.390 Å). The bond lengths C–Br are on an average 1.890 Å. Note that within the H-Bim subunit, the imidazole ring deviates by 0.735° from the benzene ring; within the acid molecule and acid dianion components, the dihedral angle between two carboxyl planes is 73.335°; and within the tetrabromoterephthalic acid subunits, the carboxyl plane and the benzene ring deviate by 84.175° and 85.190°, respectively.It was found that, in salt 2, each tetrabromoterephthalic acid dianion interacts with an acid molecule to form O4–H4⋯O2 hydrogen bonds with –OH and –COO− groups. Thus, a one-dimensional acid chain (Fig. 2b) is formed, which is quite a usual feature for this type of recognition process, and it was also observed in earlier examples. However, the interaction between adjacent chains and the resulting two-dimensional arrangement is quite fascinating. The adjacent 1D chains are interconnected by base and the water molecule via the hydrogen bonds O–H⋯O and N–H⋯O to form a two-dimensional (2D) layer (Fig. 2c). The 2D sheets in 2 are linked by the hydrogen bonds N–H⋯O, O–H⋯O and weak O–H⋯Br interactions to form supramolecular 3D network (Fig. 2d). For clarity, we presented the 2D layered structure in different colors, i.e. pink and bright green alternately, which is shown in the Fig. 2e. In addition, six types of hydrogen-bond patterns, noted as synthons II R44(18), III S5, XVI R66(20), XVII R88(40), and XV R810(52), are observed in this 3D array.
 | ||
| Fig. 2 (a) Molecular structure of 2 with atom labeling of the asymmetric unit; (b) the infinite 1D hydrogen bonding chain via O–H⋯O hydrogen bonds (the hydrogen bonds are indicated as broken lines in this and the subsequent figures); (c) perspective view of the 2D hydrogen-bonded layer; (d) the resultant 3D network; (e) the resultant 3D layer in two colours. | ||
space group (see Table 1). The asymmetric unit of 3 is comprised of two H-8-HQ+ cations, two half TBTA2− dianions and three independent H2O molecules (Fig. 3a). The tetrabromoterephthalic acid molecule is fully deprotonated with one hydroxyl proton transferred to the H-8-HQ molecule. The exocyclic bond lengths C32–C35 and C24–C41 are elongated to 1.526 Å and 1.527 Å, which are longer than the cyclic C–C bonds (average 1.387 Å). The cyclic C–N bond lengths are on an average 1.342 Å, and the C–Br bond lengths are on an average 1.890 Å. Within the acid anion components, the dihedral angle between the two benzene rings is 56.929°. Note that the dihedral angles between the pyridine ring and the benzene ring of the 8-hydroxyquinoline cation molecules are 0.727° and 1.571°, respectively.
 | ||
| Fig. 3 (a) Molecular structure of 3 with atom labeling of the asymmetric unit; (b) the infinite 1D hydrogen bonding chain via O–H⋯O hydrogen bonds (the hydrogen bonds are indicated as broken lines in this and the subsequent figures); (c) perspective view of the 2D hydrogen-bonded layer; (d) the resultant 3D layer in two colours. | ||
Analysis of the crystal structure of 3 indicates that the acid subunits and water molecules alternate, generating a one-dimensional infinite chain through the hydrogen bond O–H⋯O (Fig. 3b). Moreover, the other water molecules act as the bridge, linking the adjacent tapes. As a consequence, these 1D chains are extended to 2D sheets, and three new hydrogen-bonded patterns marked as synthons V R44(15), VI R24(8), and XVIII R1212(42) come into being. Furthermore, as shown in Fig. 3d, there exist C–H⋯Br hydrogen-bonding motifs; i.e., synthons XIX R44(26) and XX R66(23), which extend the adjacent sheets to afford a 3D supramolecular network.
 | ||
| Fig. 4 (a) Molecular structure of 4 with atom labeling of the asymmetric unit; (b) the infinite 1D hydrogen bonding chain via O–H⋯N and C–H⋯O hydrogen bonds (the hydrogen bonds are indicated as broken lines in this and the subsequent figures); (c) perspective view of the 2D hydrogen-bonded layer; (d) the resultant 3D layer in two colours. | ||
In the crystal structure of 4, the acid subunits and 5-NO2-phen molecules alternate to form a one-dimensional chain via the hydrogen bond O–H⋯N and the weak hydrogen bond C–H⋯O, as shown in Fig. 4b. These 1D chains are connected to each other to form two-dimensional layers by the O–H⋯O hydrogen bonds (synthons XIII R22(10) and XXI R810(56)), where the length is 2.700 Å. Furthermore, as viewed from the ac plane, these 2D layers are fused together to yield a 3D network through the weak hydrogen bonds C–H⋯O between acid and base components from adjacent sheets (Fig. 4d).
O, 1.205 Å) support the existence of nonionic acid moieties, indicating co-crystal formation. However, in the molecule of 4,6-dihydroxypyrimidine, depicting the intramolecular proton transfer phenomenon, the hydrogen of –OH is transferred to the nitrogen of the pyrimidine ring. The exocyclic bond length C13–C18 is elongated to 1.507 Å, longer than those of cyclic C–C bonds (average 1.392 Å). The bond lengths C–N and C–Br are on an average 1.356 Å and 1.883 Å, respectively. The angle between the calculated mean planes of the carboxylate group and its attached parent benzene ring of tetrabromoterephthalic acid is 85.311°, whereas the dihedral angle between the benzene ring and the pyrimidine ring is 38.099°.
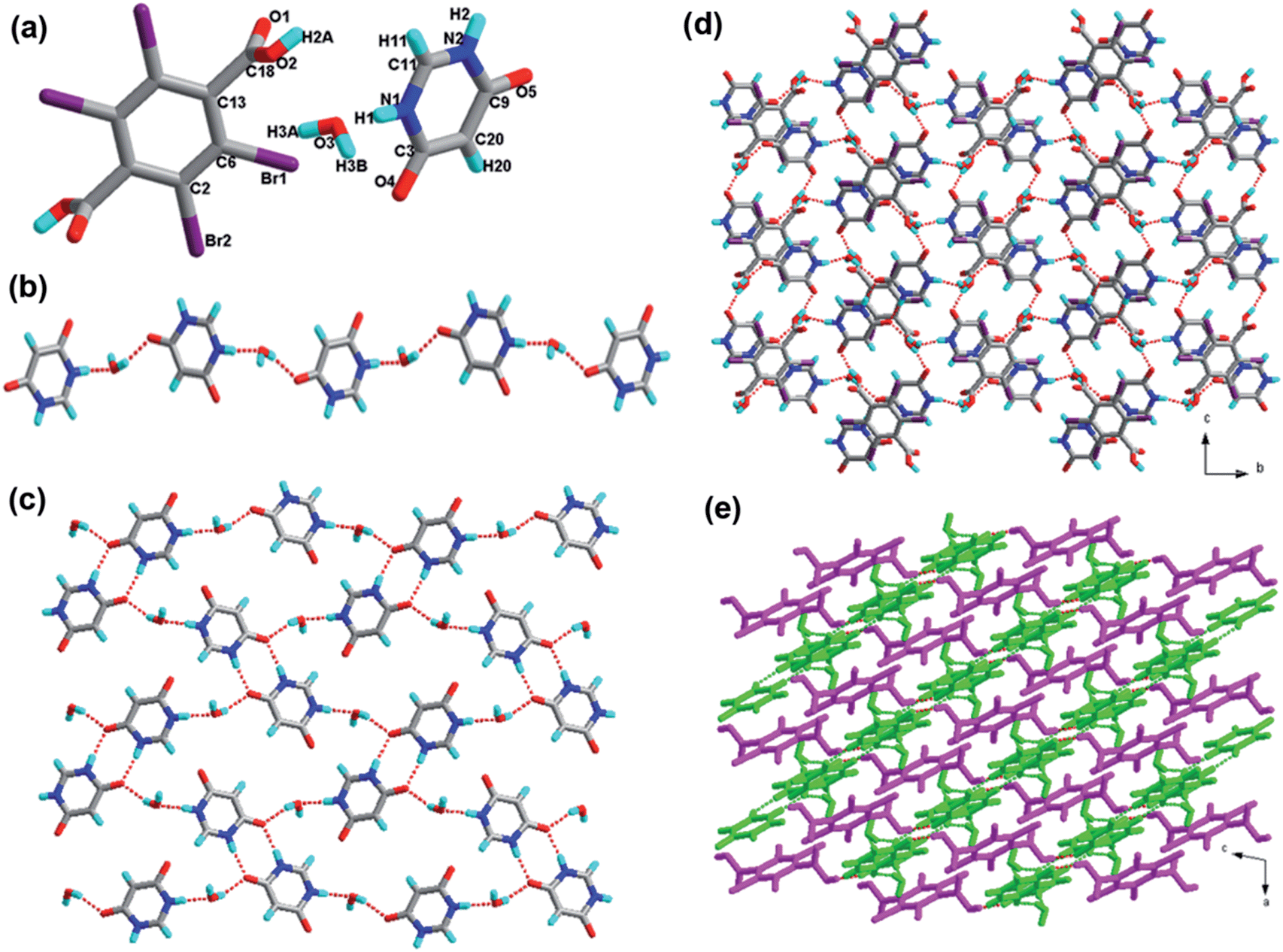 | ||
| Fig. 5 (a) Molecular structure of 5 with atom labeling of the asymmetric unit; (b) the infinite 1D hydrogen bonding chain via N–H⋯O and O–H⋯O hydrogen bonds (the hydrogen bonds are indicated as broken lines in this and the subsequent figures); (c) perspective view of the 2D hydrogen-bonded layer; (d) the resultant 3D network; (e) the resultant 3D layer in two colours. | ||
These 4,6-dihydroxypyrimidine molecule moieties and water molecules form a one-dimensional infinite hydrogen bonded chain through the O–H⋯O and N–H⋯O, which is shown in Fig. 5b. Furthermore, these 1D chains further join together via the interaction N2–H2⋯O5; moreover, synthons IX R22(8) and XXII R810(32) are observed in this structure. As a consequence, a 2D sheet network is formed through these synthons, as depicted in Fig. 5c. In addition, these 2D sheets are extended to a 3D framework (Fig. 5d) as viewed from the bc plane through the O–H⋯O bond between acid and base components from adjacent sheets. For clarity, we presented the 2D layered structure in bright green and the tetrabromoterephthalic acid moieties in pink, which is shown in Fig. 5e.
(see Table 1). As depicted in Fig. 6a, the asymmetric unit of 6 contains one H-2,4-DMI+ cation, two half H2-TBTA, and half TBTA2− anions. In 6, one tetrabromoterephthalic acid was deprotonated to form a dianionic compound. Moreover, 2,4-dimethylimidazole was protonated to obtain the monocationic compound. In the absence of hydrogen bonding and other electronic perturbations, the C–O bond lengths should be equal because of resonance. The formation of single or multiple hydrogen bonds at one oxygen atom should cause the associated C–O bond to lengthen. It is clear that the average distances for C–O (1.248 Å) in the tetrabromoterephthalic acid anion are less than the single bond C–O (1.307 Å) and greater than the double bond CO (1.205 Å) in the carboxylic acid group of the tetrabromoterephthalic acid molecule. This supports our assignment of the tetrabromoterephthalic acid dianions. The C–N bond lengths are on an average 1.348 Å. The exocyclic C–C bond lengths (average 1.513 Å) are longer than cyclic C–C bond lengths (average 1.388 Å). The exocyclic C–Br bond lengths are on an average 1.884 Å. The angles between the tetrabromoterephthalic acid molecules and tetrabromoterephthalic acid anion are 82.948° and 13.103°, respectively.
 | ||
| Fig. 6 (a) Molecular structure of 6 with atom labeling of the asymmetric unit; (b) the infinite 1D hydrogen bonding chain via O–H⋯O hydrogen bonds (the hydrogen bonds are indicated as broken lines in this and the subsequent figures); (c) perspective view of the 2D hydrogen-bonded layer; (d) the resultant 3D layer in two colours. | ||
These tetrabromoterephthalic acid dianion and molecular moieties form a linear infinite hydrogen bonded chain with adjacent –COOH and –COO− groups of neighboring moieties with the anionic and molecular moieties arranged alternately, running parallel to the a-axis as shown in Fig. 6b. Furthermore, the adjacent 1D chains were connected via the hydrogen bond O–H⋯O. On the basis of these connection modes, these 1D chains are linked to generate a 2D layer structure (Fig. 6c). To further understand the structure of 6, the 3D network was carried out. As depicted in Fig. 6d, the 2D sheets were displayed in pink and the H-2,4-DMI molecules were shown in bright green. They connected through the N–H⋯O to generate a 3D supramolecular structure. In addition, four types of synthons, noted as X R22(12), XXIII R64(32), XXIV R66(29) and XXV R66(39) are observed in this 3D array.
(see Table 1). The asymmetric unit of the structure for compound 7 is composed of one H-3,5-DMP+ cation and half TBTA2− anion. As shown in Fig. 7a, 3,5-dimethylpyrazole accepts one hydrogen from half a tetrabromoterephthalic acid molecule. The acidic –COOH hydrogen on tetrabromoterephthalic acid has been transferred to the basic –N– moiety of 3,5-dimethylpyrazole. In the absence of hydrogen bonding and other electronic perturbations, the C–O bond lengths should be almost equal in the molecule of tetrabromoterephthalic acid (O1–C4, 1.243 Å; O2–C4, 1.244 Å) because of resonance. The exocyclic C–C bond lengths (C4–C8, 1.534 Å) are longer than cyclic C–C bond lengths (average 1.389 Å). The exocyclic C–Br bond lengths are on an average 1.891 Å, whereas the C–N bond lengths are on an average 1.338 Å. The dihedral angle between the benzene ring of tetrabromoterephthalic acid anion and the pyrazole ring of the 3,5-dimethylpyrazole anion is 37.206°.
 | ||
| Fig. 7 (a) Molecular structure of 7 with atom labeling of the asymmetric unit; (b) the infinite 1D hydrogen bonding chain via N–H⋯O and C–H⋯O hydrogen bonds (the hydrogen bonds are indicated as broken lines in this and the subsequent figures); (c) perspective view of the 2D hydrogen-bonded layer; (d) the resultant 3D layer in two colours. | ||
Analysis of the crystal of 7 indicates that the acid subunits and base molecules alternate, generating a one-dimensional infinite chain through the hydrogen bond N–H⋯O and the weak interaction C–H⋯O (Fig. 7b). Furthermore, the adjacent 1D chains are connected to each other through C–H⋯O. As a consequence, these 1D chains are extended to a 2D sheet (shown in Fig. 7c), and three new hydrogen-bonded patterns marked as XXVI R65(25) come into being, Significantly, as shown in Fig. 7d, there exist the weak hydrogen bonds C–H⋯O. These 2D layers extend the adjacent sheets to afford a 3D supramolecular network. Synthons XI R24(8), XII R44(18), XXVII R44(28) and XXVIII R44(30) are formed properly and shown in Scheme 2 and ESI.†
space group, and the asymmetric unit is found to be composed of one H-4-CNpy+ cation, half H2-TBTA and half TBTA2− anion. As shown in Fig. 8a, the state of the carboxylic moiety (neutral or ionic) can be found through the C–O and CO bond distances. The lengths of two C–O bonds for the tetrabromoterephthalic acid molecule are obviously different, and they are 1.304 Å for C–OH and 1.195 Å for CO. The two C–O bonds for tetrabromoterephthalic acid anion are similar (1.207 Å and 1.303 Å), showing that one acid is neutral and the other is fully deprotonated. The tetrabromoterephthalic acid molecule is fully deprotonated with one hydroxyl proton transferred to the 4-cyanopyridine. The exocyclic bond lengths C3–C8, C12–C24 and C13–C27 are elongated to 1.510 Å, 1.527 Å and 1.439 Å, respectively, which is longer than those of the cyclic C–C bonds (average 1.380 Å). The cyclic bond lengths C–N are on an average 1.327 Å, and the exocyclic bond lengths C–Br are on an average 1.883 Å. The dihedral angle between the two benzene rings of the acid subunits is 75.865°. In addition, the dihedral angles between the pyridine ring and the two benzene rings are 0.828° and 75.282°, respectively.
 | ||
| Fig. 8 (a) Molecular structure of 8 with atom labeling of the asymmetric unit; (b) the infinite 1D hydrogen bonding chain via C–H⋯O, C–H⋯N and N–H⋯O hydrogen bonds (the hydrogen bonds are indicated as broken lines in this and the subsequent figures); (c) perspective view of the 2D hydrogen-bonded layer; (d) the resultant 3D layer in two colours. | ||
To further understand the structure of 8, the one-dimensional chain was investigated. In 8, the adjacent 4-cyanopyridine molecules were connected to form a new hydrogen-bonded ring with the graph-set of XIII R22(10). Moreover, the synthons XIII R22(10) and H2-TBTA molecules alternate, generating a 1D infinite chain via the hydrogen bond C–H⋯O (Fig. 8b). Tetrabromoterephthalic acid anions act as bridges, linking the adjacent 1D chains through O–H⋯O. As a consequence, these 1D chains are extended to a 2D sheet, and new hydrogen-bonded patterns marked as synthons XXIX R66(30), XXX R46(30), XXXI R68(26) come into being. Such neighboring 2D layers are further cross-linked via the N1–H1⋯O1 hydrogen bond, generating a 3D structure along a-axis. In Fig. 8d, the 2D layers were depicted in different colors; therefore, the connection between each was displayed more clearly.
Thermal stability analysis
All complexes 1–8 are stable in air and can maintain their structural integrity at ambient conditions for a long time. In order to examine the thermal stability of all complexes, TGA and DSC were carried out between room temperature and 900 °C in a nitrogen atmosphere. The DSC traces and TGA data for the crystals are presented in the ESI.† TGA experiments were implemented to investigate their thermal stability. The behaviors of the eight complexes are depicted in Fig. 9. As for complex 6, the TGA results indicate that they remain intact until 218 °C, and then there is a sharp weight loss ending at 260 °C (peaks: 225.1 °C for crystal 6). The weight loss of complex 6 is up to 99.96% at 260 °C. The TGA curves of 2, 7 and 8 indicate that there are two consecutive weight losses of the three samples. Complex 2 decomposes from 100 °C to 258 °C (peaking at 256 °C), whereas 7 and 8 are more stable than the compound 1 and when it comes to 140 °C and 160 °C, respectively, the decomposition of the framework begins (peaking at 237 °C, 351 °C and 198 °C, 355 °C, respectively). As for 2, the first weight loss of 3.25% from 100 to 110 °C (calculated: 2.91%) corresponds to the loss of two water molecules per formula. The second weight loss of 96.87% (calculated: 97.09%) can be detected from 220 to 258 °C, which is due to the decomposition of acid and base molecules. Compared with complex 1, the TGA measurement of 7 shows a weight loss of 30.51% in the temperature range 110 °C to 155 °C, which corresponds to the loss of a 3,5-dimethylpyrazole molecule, and the second weight loss represents the loss of acidic components (calculated: 71.47%, found: 80.09%). The TGA measurement of 8 indicates that the complex does not melt and is stable up to 145 °C, and at this temperature the crystal begins to decompose. The ligand 4-cyanopyridine and tetrabromoterephthalic acid molecule decompose at 145–368 °C with two peaks at 198 °C and 355 °C. As for 5, the first weight loss of 5.13% (calculated: 4.85%) occurs from 95 to 170 °C, which corresponds to the loss of two water molecules per formula, and the second weight loss of 27.64% occurs from 195 to 270 °C, and the third weight loss of 61.30% occurs from 270 to 338 °C. As for 1, the first weight loss of 14.66% occurs from 155 to 210 °C, the second weight loss of 19.41% occurs from 210 to 245 °C, and the third weight loss of 65.53% occurs from 245 to 326 °C. For complex 4, two consecutive weight losses occur in the temperature range of 167–610 °C (peaking at 235 °C and 519 °C, respectively). The TGA curve of complex 3 indicates that it has three trends of decomposition. The sample is stable up to 78 °C, and at this temperature, it starts to melt and decompose. The first mass loss of 7.22% (calculated: 6.54%) corresponds to the loss of three water molecules per formula, the second mass loss represented the base molecules, and the third mass loss of 55.69% corresponds to the acid molecules (calculated: 58.23%). Moreover, the theoretical value and experimental value differ within a reasonable range. Broadly speaking, the eight frameworks have remarkable thermal stability. | ||
| Fig. 9 The TGA curves of complexes 1–8. | ||
Conclusions
The self-assembly of designed molecules containing certain kinds of synthons has stimulated new efforts in material science. Such crystal manipulations, often known as crystal engineering, are performed to yield new structural molecular solids. The successful preparation of the eight organic acid–base adducts in this paper provides novel 3D network structures by using suitable flexible ligands. These structures contribute to the extensive research into the occurrence of tetrabromoterephthalic acid compound motifs in organic solids.From this study, it can be seen that tetrabromoterephthalic acid will form organic solids with the basic molecules. With the exception of compounds 4 and 5, the other six complexes are formed from salts by the proton transfer process. Under normal conditions, the proton of the carboxylic acid transfers to the nitrogen atoms in the basic compounds. Crystals 4 and 5 formed cocrystals and maintain the original molecular structure. The strong intermolecular N–H⋯O and O–H⋯O hydrogen bonds generally exist in these structures. These interactions are responsible for the high-yielding supramolecular assembly of tetrabromoterephthalic acid and N-heterocycles into organic solids with desired connectivities. In addition, the weak interactions C–H⋯O, C–H⋯Br and N–H⋯Br were observed based upon their geometric preferences and showed equal importance to the strong hydrogen bonds in these structures. There are also π–π stacking interactions in the compound 1, in which the closest separation between centers of aromatic rings is 3.644 Å. More importantly, the bromine atom played a vital important role in constructing these crystal structures through the C–H⋯Br and N–H⋯Br hydrogen bond interactions.
It should be noted that although these N-heterocycles, such as H-BTAH, 4-CNpy, H-Bim, and 4,6-DHP ligands, present a planar space configuration: 8-HQ, 3,5-DMP, 2,4-DMI display a three-dimensional space configuration due to the –OH, –CH3 groups, and all crystal structures containing these ligands are 3D supramolecular networks. These results are mainly due to the existence of the large number of hydrogen bonds (O–H⋯O, O–H⋯N, N–H⋯O, N–H⋯N, C–H⋯O, C–H⋯N, C–H⋯Br, N–H⋯Br) and Br⋯Br interactions present in these structures. In future studies, we will continue to research the halogen compounds and to discover the C–H⋯halogen and halogen⋯halogen interactions in the crystal construction.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 51372125, 21371105, and 21203106), and the Scientific and Technical Development Project of Qingdao (no. 13-1-4-184-jch).Notes and references
- (a) K. Biradha, A. Nangia, G. R. Desiraju, C. J. Carrell and H. L. Carrell, J. Mater. Chem., 1997, 7, 1111–11221 RSC; (b) V. R. Thalladi, S. Brasselet, D. Bläser, R. Boese, J. Zyss, A. Nangia and G. R. Desiraju, Chem. Commun., 1997, 19, 1841–1842 RSC; (c) S. S. Kuduva, D. C. Craig, A. Nangia and G. R. Desiraju, J. Am. Chem. Soc., 1999, 121, 1936–1944 CrossRef CAS; (d) G. A. Hembury, V. V. Borovkov and Y. Inoue, Chem. Rev., 2008, 108, 1–73 CrossRef CAS PubMed; (e) H. Cho, L. Widanapathirana and Y. Zhao, J. Am. Chem. Soc., 2011, 133, 141–147 CrossRef CAS PubMed; (f) Q. P. Duan, Y. Cao, Y. Li, X. Y. Hu, T. X. Xiao, C. Lin, Y. Pan and L. Y. Wang, J. Am. Chem. Soc., 2013, 135, 10542–10549 CrossRef CAS PubMed; (g) X. F. Ji, J. Y. Li, X. Z. Yan and F. H. Huang, J. Am. Chem. Soc., 2013, 135, 74–77 CrossRef CAS PubMed; (h) F. Awwadi, S. F. Haddad, R. D. Willett and B. Twamley, Cryst. Growth Des., 2010, 10, 158–164 CrossRef CAS; (i) H. P. Zhou, J. H. Yin, L. X. Zheng, P. Wang, F. Y. Hao, W. Q. Geng, X. P. Gan, G. Y. Xu, J. Y. Wu, Y. P. Tian, X. T. Tao, M. H. Jiang and Y. H. Kan, Cryst. Growth Des., 2009, 9, 3789–3798 CrossRef CAS; (j) G. R. Desiraju, J. Mol. Struct., 2003, 656, 5–15 CrossRef CAS; (k) F. T. Martins, N. Paparidis, A. C. Doriguetto and J. Ellena, Cryst. Growth Des., 2009, 9, 5283–5292 CrossRef CAS.
- (a) S. S. Kuduva, D. C. Craig, A. Nangia and G. R. Desiraju, J. Am. Chem. Soc., 1999, 121, 1936–1944 CrossRef CAS; (b) F. Awwadi, S. F. Haddad, R. D. Willett and B. Twamley, Cryst. Growth Des., 2010, 10, 158–164 CrossRef CAS.
- (a) G. A. Hembury, V. V. Borovkov and Y. Inoue, Chem. Rev., 2008, 108, 1–73 CrossRef CAS PubMed; (b) X. F. Ji, J. Y. Li, X. Z. Yan and F. H. Huang, J. Am. Chem. Soc., 2013, 135, 74–77 CrossRef CAS PubMed; (c) C. Y. Wang, J. G. Wang, P. Z. Li, J. K. Gao, S. Y. Tan, W. W. Xiong, B. L. Hu, P. S. Lee, Y. L. Zhao and Q. C. Zhang, Chem.–Asian J., 2014, 9, 779–783 CrossRef CAS PubMed; (d) G. Li, Y. C. Wu, J. K. Gao, C. Y. Wang, J. B. Li, H. C. Zhang, Y. Zhao, Y. L. Zhao and Q. C. Zhang, J. Am. Chem. Soc., 2012, 134, 20298–20301 CrossRef CAS PubMed; (e) J. F. Zhao, G. Li, C. Y. Wang, W. Q. Chen, S. C. J. Loo and Q. C. Zhang, RSC Adv., 2013, 3, 9653–9657 RSC; (f) G. Li, Y. C. Wu, J. K. Gao, J. B. Li, Y. Zhao and Q. C. Zhang, Chem.–Asian J., 2013, 8, 1574–1578 CrossRef CAS PubMed.
- (a) Q. P. Duan, Y. Cao, Y. Li, X. Y. Hu, T. X. Xiao, C. Lin, Y. Pan and L. Y. Wang, J. Am. Chem. Soc., 2013, 135, 10542–10549 CrossRef CAS PubMed; (b) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- (a) A. S. Singh, B. Y. Chen, Y. S. Wen, C. Tsai and S. S. Sun, Org. Lett., 2009, 11, 1867–1870 CrossRef CAS PubMed; (b) L. Wang, L. Zhao, L. Y. Xu, R. X. Chen and Y. Yang, CrystEngComm, 2012, 14, 6998–7008 RSC; (c) L. Wang, R. F. Xue, L. Y. Xu, X. Lu, R. X. Chen and X. T. Tao, Sci. China: Chem., 2012, 55, 1228–1235 CrossRef CAS; (d) B. Yang, J. C. Xiao, J. I. Wong, J. Guo, Y. C. Wu, L. J. Ong, L. L. Lao, F. Boey, H. Zhang, H. Y. Yang and Q. C. Zhang, J. Phys. Chem. C, 2011, 115, 7924–7927 CrossRef CAS; (e) Z. Q. Lin, P. J. Sun, Y. Y. Tay, J. Liang, Y. Liu, N. E. Shi, L. H. Xie, M. D. Yi, Y. Qian, Q. L. Fan, H. Zhang, H. H. Hng, J. Ma, Q. C. Zhang and W. Huang, ACS Nano, 2012, 6, 5309–5319 CrossRef CAS PubMed; (f) J. C. Xiao, B. Yang, J. I. Wong, Y. Liu, F. X. Wei, K. J. Tan, X. Teng, Y. C. Wu, L. Huang, C. Kloc, F. Boey, J. Ma, H. Zhang, H. Y. Yang and Q. C. Zhang, Org. Lett., 2011, 13, 3004–3007 CrossRef CAS PubMed; (g) J. C. Xiao, Z. Y. Yin, Y. C. Wu, J. Guo, Y. H. Cheng, H. Li, Y. Z. Huang, Q. Zhang, J. Ma, F. Boey, H. Zhang and Q. C. Zhang, Small, 2011, 7, 1242–1246 CrossRef CAS PubMed; (h) Y. Liu, F. Boey, L. L. Lao, H. Zhang, X. G. Liu and Q. C. Zhang, Chem.–Asian J., 2011, 6, 1004–1006 CrossRef CAS PubMed.
- (a) M. Yamada, R. Kanazawa and F. Hamada, CrystEngComm, 2014, 16, 2605–2614 RSC; (b) I. Saraogi, V. G. Vijay, S. Das, K. Sekar and T. N. Guru Row, Cryst. Eng., 2003, 6, 69–77 CrossRef CAS; (c) C. B. Aakeröy and A. M. Bratty, Aust. J. Chem., 2001, 54, 409–421 CrossRef; (d) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed; (e) M. Morgan Conn and J. Rebek Jr, Chem. Rev., 1997, 97, 1647–1668 CrossRef; (f) S. Yagai, T. Nakajima, K. Kishikawa, S. Kohmoto, T. Karatsu and A. Kitamura, J. Am. Chem. Soc., 2005, 127, 11134–11139 CrossRef CAS PubMed; (g) H. B. Yang, K. Ghosh, B. H. Northrop, Y. R. Zheng, M. M. Lyndon, D. C. Muddiman and P. J. Stang, J. Am. Chem. Soc., 2007, 129, 14187–14189 CrossRef CAS PubMed; (h) F. F. Awwadi, R. D. Willett and B. Twamley, Cryst. Growth Des., 2007, 7, 624–632 CrossRef CAS.
- (a) N. Stanley, V. Sethuraman, P. Thomas Muthiah, P. Luger and M. Weber, Cryst. Growth Des., 2002, 2, 631–635 CrossRef CAS; (b) S. Baskar Raj, P. Thomas Muthiah, G. Bocelli, R. Ollá and A. Cantoni, Cryst. Growth Des., 2003, 3, 567–571 CrossRef; (c) R. Thaimattam, D. S. Reddy, F. Xue, T. C. W. Mak, A. Nangia and G. R. Desiraju, J. Chem. Soc., Perkin Trans. 2, 1998, 1783–1789 RSC; (d) L. Wang, L. Zhao, W. Liu, R. X. Chen, Y. X. Gu and Y. Yang, Sci. China: Chem., 2012, 55, 2381–2387 CrossRef CAS; (e) L. Wang, L. Zhao, R. F. Xue, X. Lu, Y. H. Wen and Y. Yang, Sci. China: Chem., 2012, 55, 2515–2522 CrossRef CAS.
- (a) J. N. Moorthy, R. Natarajan, P. Mal and P. Venugopalan, J. Am. Chem. Soc., 2002, 124, 6530–6531 CrossRef CAS PubMed; (b) L. Wang, L. Zhao, M. Liu, F. Q. Liu, Q. Xiao and Z. Hu, Sci. China: Chem., 2012, 55, 2523–2531 CrossRef CAS; (c) L. Wang, L. Zhao, M. Liu, R. X. Chen, Y. Yang and Y. X. Gu, Sci. China: Chem., 2012, 55, 2115–2122 CrossRef CAS.
- (a) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS PubMed; (b) S. Berski, Z. Ciunik, K. Drabent, Z. Latajka and J. Panek, J. Phys. Chem. B, 2004, 108, 12327–12332 CrossRef CAS; (c) M. L. Tong, Z. J. Lin, W. Lin, S. L. Zheng and X. M. Chen, Cryst. Growth Des., 2002, 2, 443–448 CrossRef CAS; (d) K. Dziubek, M. Podsiadlo and A. Katrusiak, J. Phys. Chem. B, 2009, 113, 13195–13201 CrossRef CAS PubMed; (e) M. Yamada, R. Kanazawa and F. Hamada, CrystEngComm, 2014, 16, 2605–2614 RSC; (f) F. F. Awwadi, R. D. Willett and B. Twamley, Cryst. Growth Des., 2007, 7, 624–632 CrossRef CAS; (g) D. Cao, M. Hong, A. K. Blackburn, Z. C. Liu, J. M. Holcroft and J. F. Stoddart, Chem. Sci., 2014, 5, 4242–4248 RSC.
- F. Guthrie, J. Chem. Soc., 1863, 16, 239–244 RSC.
- (a) R. Glaser, N. J. Chen, H. Wu, N. Knotts and M. Kaupp, J. Am. Chem. Soc., 2004, 126, 4412–4419 CrossRef CAS PubMed; (b) L. Wang, L. Y. Xu, R. Xue, X. Lu, R. X. Chen and X. Tao, Sci. China: Chem., 2012, 55, 138–144 CrossRef CAS; (c) P. T. Pham and M. M. Bader, Cryst. Growth Des., 2014, 14, 916–922 CrossRef CAS.
- (a) M. Fourmigué and P. Batail, Chem. Rev., 2004, 104, 5379–5418 CrossRef PubMed; (b) N. S. Goroff, S. M. Curtis, J. A. Webb, F. W. Foeler and J. W. Lauher, Org. Lett., 2005, 7, 1891–1893 CrossRef CAS PubMed; (c) Md. B. Zaman, K. A. Udachin and J. A. Ripmeester, Cryst. Growth Des., 2004, 4, 585–589 CrossRef CAS; (d) F. Zordan and L. Brammer, Cryst. Growth Des., 2006, 6, 1374–1379 CrossRef CAS; (e) R. D. Willett, F. Awwadi and R. Butcher, Cryst. Growth Des., 2003, 3, 301–311 CrossRef CAS; (f) K. A. Kounavi, M. J. Manos, E. E. Moushi, A. A. Kitos, C. Papatriantafyllopoulou, A. J. Tasiopoulos and V. Nastopoulos, Cryst. Growth Des., 2012, 12, 429–444 CrossRef CAS.
- (a) F. Zapata, A. Caballero, N. G. White, T. D. W. Claridge, P. J. Costa, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2012, 134, 11533–11541 CrossRef CAS PubMed; (b) H. Y. Gao, X. R. Zhao, H. Wang, X. Pang and W. J. Jin, Cryst. Growth Des., 2012, 12, 4377–4387 CrossRef CAS; (c) J. Viger-Gravel, S. Leclerc, I. Korobkov and D. L. Bryce, CrystEngComm, 2013, 15, 3168–3177 RSC; (d) T. J. Mooibroek and P. Gamez, CrystEngComm, 2013, 15, 4565–4570 RSC; (e) X. Y. Yang, F. Wang, Q. X. Chen, L. Y. Wang and Z. Q. Wang, Chin. Sci. Bull., 2007, 52, 1856–1859 CrossRef CAS; (f) P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS PubMed.
- (a) C. P. Li, J. Chen and M. Du, CrystEngComm, 2010, 12, 4392–4402 RSC; (b) C. P. Li, Y. L. Tian and Y. M. Guo, Inorg. Chem. Commun., 2008, 11, 1405–1408 CrossRef CAS; (c) P. G. Waddell, J. O. S. Hulse and J. M. Cole, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2011, 67, o255–o258 CAS; (d) Z. Hulvey, E. Ayala and A. K. Cheetham, Z. Anorg. Allg. Chem., 2009, 635, 1753–1757 CrossRef CAS; (e) Z. Hulvey, J. D. Furman, S. A. Turner, M. Tang and A. K. Cheetham, Cryst. Growth Des., 2010, 10, 2041–2043 CrossRef CAS; (f) Y. Ma, X. P. Chen, D. Cao, S. P. Yan and D. Z. Liao, Sci. China, Ser. B: Chem., 2009, 52, 1438–1443 CrossRef CAS; (g) C. Seidel, R. Ahlers and U. Ruschewitz, Cryst. Growth Des., 2011, 11, 5053–5063 CrossRef CAS; (h) Z. Hulvey, E. Ayala, J. D. Furman, P. M. Forster and A. K. Cheetham, Cryst. Growth Des., 2009, 9, 4759–4765 CrossRef CAS; (i) A. Orthaber, C. Seidel, F. Belaj, J. H. Albering, R. Pietschnig and U. Ruschewitz, Inorg. Chem., 2010, 49, 9350–9357 CrossRef CAS PubMed; (j) R. Kitaura, F. Iwahori, R. Matsuda, S. Kitagawa, Y. Kubota, M. Takata and T. C. Kobayashi, Inorg. Chem., 2004, 43, 6522–6524 CrossRef CAS PubMed; (k) L. Wang, L. Zhao, Y. J. Hu, W. Q. Wang, R. X. Chen and Y. Yang, CrystEngComm, 2013, 15, 2835–2852 RSC; (l) L. Wang, Y. J. Hu, W. Q. Wang, F. Q. Liu and K. K. Huang, CrystEngComm, 2014, 16, 4142–4161 RSC.
- (a) G. M. Sheldrick, SHELXS-97, Program for the Solution of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, SHELXL-97, Programs for X-ray Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: IR, TGA, DSC data and synthons XIV–XXXI about this work. CCDC 1016043–1016050 For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra08452g |
| This journal is © The Royal Society of Chemistry 2014 |