
Mesopattern of immobilised bone morphogenetic protein-2 created by microcontact printing and dip-pen nanolithography influence C2C12 cell fate†
Abstract
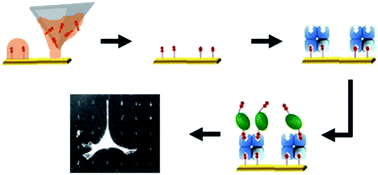
Dip-pen nanolithography and microcontact printing were used to fabricate mesopatterned substrates for cell differentiation experiments. A biotin–thiol was patterned on gold substrates and subsequently functionalised with streptavidin and biotinylated bone morphogenetic protein-2 (BMP-2). The feasibility of mesopatterned substrates containing immobilised BMP-2 was proven by obtaining similar differentiation outcomes compared to the growth factor in solution. Therefore, these substrates might be suitable for replacing conventional experiments with BMP-2 in solution.
 Please wait while we load your content...
Please wait while we load your content...

