
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The aqueous N-phosphorylation and N-thiophosphorylation of aminonucleosides†
Louis P. Conway,
Richard J. Delley,
Jonathan Neville,
Gemma R. Freeman,
Hannah J. Maple,
Vincent Chan,
Alexander J. Hall and
David R. W. Hodgson*
Department of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: d.r.w.hodgson@durham.ac.uk; Tel: +44 (0)191 33 42123
First published on 27th August 2014
Abstract
We demonstrate N-phosphorylation and N-thiophosphorylation of unprotected aminonucleosides in aqueous media. N-Phosphorylations using phosphoric chloride and N-thiophosphorylations using thiophosphoryl chloride were explored as functions of pH using 5′-amino-5′-deoxyguanosine as substrate. These reagents were compared to phosphodichloridate and thiophosphodichloridate ions, and the methodology was applied to other aminonucleosides. S-Alkylations of the nucleoside N-thiophosphoramidates were investigated as functions of pH and alkylating agent.
Introduction
Nucleoside phosphates are ubiquitous in living systems, with nucleic acids, adenosine triphosphate, and nucleotide sugars being key examples. The synthesis of analogues of these compounds in which the phosphate groups have been modified has been widely explored.1–4 Phosphate-modified nucleotides have a number of applications, including investigations of the roles of phosphate in the binding of nucleoside monophosphates to enzymes.5,6 Modified nucleosides and nucleotides also offer reactive functionalites for bioconjugation.7–10 Standard methods for the synthesis of nucleoside phosphates, such as the Poulter11 and Yoshikawa12 syntheses, are frequently cumbersome, and require rigorously dry conditions and laborious purification. The use of the phosphoramidite method, while highly efficient for the synthesis of oligonucleotides when the protected nucleoside phosphoramidites are commercially available, becomes more laborious and requires global protection and rigorously dry conditions where the de novo syntheses of non-standard phosphoramidites are required.13These problems motivated us to find a simple method for the synthesis of analogues of nucleoside monophosphates and phosphodiesters, building on our previous work in the area.14–16 Whilst previous syntheses of the 5′-N-phosphoramidates of guanosine17 and other nucleosides5,6,18 have been reported, these methods suffer from similar drawbacks to the Yoshikawa synthesis. The issues centre on poor nucleoside solubility and the need for dry conditions. In contrast, we find that the aqueous phosphorylation method is more convenient, where a nucleophilic aminonucleoside allowed phosphorylation to be carried out in water, with high conversion, and good N-selectivity.14,15 The methodology was extended to the use of thiophosphoryl chloride as a (thio)phosphorylation agent on a series of generic amines, with the advantage that the thiophosphoramidates thus formed can then be S-alkylated to produce analogues of phosphodiester systems.19
Here we seek to explore, expand and optimise the aqueous phosphorylations and thiophosphorylations of aminonucleosides through pH control, and to compare phosphorylating and thiophosphorylating agents. We have also previously reported on the hydrolysis kinetics of phosphodichloridate (Cl2OPO−) and thiophosphodichloridate (Cl2SPO−) ions,20 and here we assess their suitability as phosphorylating agents, compared to their counterparts POCl3 and PSCl3.
Results and discussion
pH control and optimisation
5′-Amino-5′-deoxyguanosine 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 was used as a model substrate to determine pH optima for phosphorylation procedures. The 5′-amino-5′-deoxyguanosine 3 was dissolved in water and adjusted to a predetermined pH value with potassium hydroxide solution. A single equivalent of the phosphorylating agent in acetonitrile was then added slowly to the solution whilst the pH was kept constant using an autotitrator to add potassium hydroxide solution as required.
21 was used as a model substrate to determine pH optima for phosphorylation procedures. The 5′-amino-5′-deoxyguanosine 3 was dissolved in water and adjusted to a predetermined pH value with potassium hydroxide solution. A single equivalent of the phosphorylating agent in acetonitrile was then added slowly to the solution whilst the pH was kept constant using an autotitrator to add potassium hydroxide solution as required.
The 5′-amino-5′-deoxyguanosine 3 was either phosphorylated or thiophosphorylated using one of phosphoryl chloride 1(O), thiophosphoryl chloride 1(S), potassium phosphodichloridate 2(O) or potassium thiophosphodichloridate 2(S) at pH values of 11, 11.5, 12, and 12.5 (Fig. 1). After addition of the phosphorylating/thiophosphorylating agent, the conversion levels were determined by 31P NMR spectroscopy. The N-phosphoramidate and N-thiophosphoramidate monoesters are unstable, except at high pH,14,22,23 making them difficult to isolate, however, 5′-amino-5′-deoxyguanosine 5′-N-phosphoramidate has been isolated and characterised by 31P NMR and 1H NMR spectroscopies, and these data were used to corroborate the observed signals.14 The presence of the N-thiophosphoramidate was confirmed by trapping this reactive species through S-alkylation and isolating and characterising the stable adduct, in a manner analogous to our earlier report.22
 | ||
| Fig. 1 The phosphosphorylating agents 1 and 2 used in this study, and the model substrate, 5′-amino-5′-deoxyguanosine 3. | ||
Apart from the desired N-phosphoramidate 5 and N-thiophosphoramidate 8, we also observed the formation of bis-aminolysis side products 6 and 9, in which the phosphorylating agent was attacked by two equivalents of amine, and inorganic phosphate 7 and thiophosphate 10, which result from the breakdown of the phosphorylating agents. These results mimic those we have seen previously for a range of generic amines 4 (Scheme 1).19,22
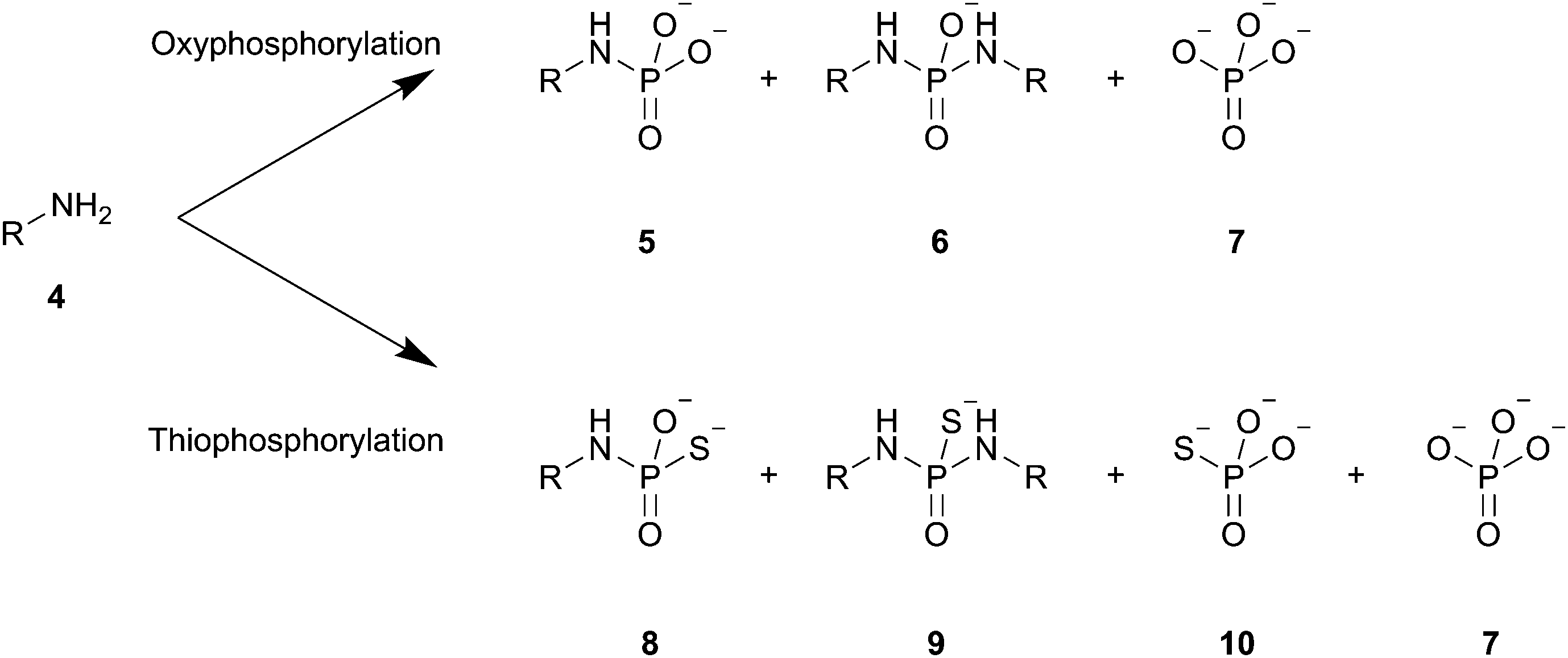 | ||
| Scheme 1 The observed products and side-products of the phosphorylation and thiophosphorylation reactions. R = 5′-deoxyguanosyl. | ||
The results show optima at pH 12 for each of the four (thio)phosphorylating agents 1(O), 1(S), 2(O) and 2(S) (Fig. 2). The optima were especially pronounced for the thiophosphorylation reactions; the greatest levels of conversion were achieved using the thiophosphodichloridate ion 2(S), with a maximum of 97% conversion to thiophosphoramidate 8 at pH 12, as opposed to 87% at pH 11. The thiophosphorylation reactions with thiophosphoryl chloride 1(S) reflect the same trend, with a maximum of 93% at pH 12, and minimum of 79% at pH 11.
 | ||
| Fig. 2 The results of the N-phosphorylations and N-thiophosphorylations of 5′-amino-5′-deoxyguanosine. Notation: (red) product; (green) inorganic thiophosphate; (blue) inorganic phosphate; (yellow) bis product; (black) other. | ||
The effect of pH on the oxyphosphorylation appears to be less pronounced. The greatest level of conversion was again achieved using the phosphodichloridate ion 2(O) at pH 12, with 83% to 76% being observed over the tested pH range. The reactions with phosphoryl chloride 1(O) were marginally poorer with conversions in the range 80% to 72%. Analysis of the by-products revealed a clear advantage to the use of the phosphodichloridate 2(O), namely, the lack of formation of bis aminolysis product 6.
Considering the structure of the substrate, 5′-amino-5′-deoxyguanosine 3 and the (thio)phosphorylating agents, several factors could account for these trends. High pHs are required to stabilise the (thio)phosphoramidate products, however, they also serve to solubilise the 5′-amino-5′-deoxyguanosine 3 substrate through ionisation of the N–H of the guanine. At higher pHs, the ionisation of the cis-diol functionality could contribute to a change in reaction outcome (pKa ∼ 12.5).24 In the case of the dichloridate ions 2(O) and 2(S), hydroxide-promoted hydrolysis of the (thio)phosphorylating agent is unlikely in light of our previous studies upon these species, which reveal flat pH–kobs profiles.20 With trichlorides 1(O) and 1(S), their enhanced reactivity could render them more susceptible to hydrolysis, however, their hydrolysis products are the dichloridates 2(O) and 2(S), which permit additional opportunity for nucleophilic attack (loss of the “third” chloride is likely to be unselective towards the nature of the nucleophile, and thus would reflect nucleophile concentration, where water is clearly the most abundant entity). Solubility phenomena and the partitioning of reaction partners between different phases could also influence reaction outcome significantly, however, the effects of these properties are difficult to predict.
Synthesis of other aminonucleosides
Having established a single pH optimum across all four phosphorylating agents 1(O), 1(S), 2(O) and 2(S) using the model substrate 5′-amino-5′-deoxyguanosine 3, we applied the methodology to other nucleosides. Four aminonucleosides were prepared to test the generality of our finding: 5′-amino-5′-deoxyadenosine 11, 3′-amino-3′-deoxythymidine 12, 5′-amino-5′-deoxyuridine 15 and 5′-amino-5′-deoxycytidine 20. The dihydrochloride salt of 5′-amino-5′-deoxyadenosine 11 was synthesised according to a literature method.25 The 3′-amino-3′-deoxy analogue of thymidine 12 was synthesised starting from the commercially available 3′-azido-3′-deoxythymidine (AZT), based on a procedure by Pastor-Anglada et al.26 and was precipitated as a hydrochloride salt 12·HCl.5′-Amino-5′-deoxyuridine was formed by adapting a single step procedure developed by Hata et al. to convert uridine to 5′-azido-5′-deoxyuridine 14,27 which was reduced to the desired amine using triphenylphosphine,26 and isolated as its hydrochloride salt 15·HCl (Scheme 2).
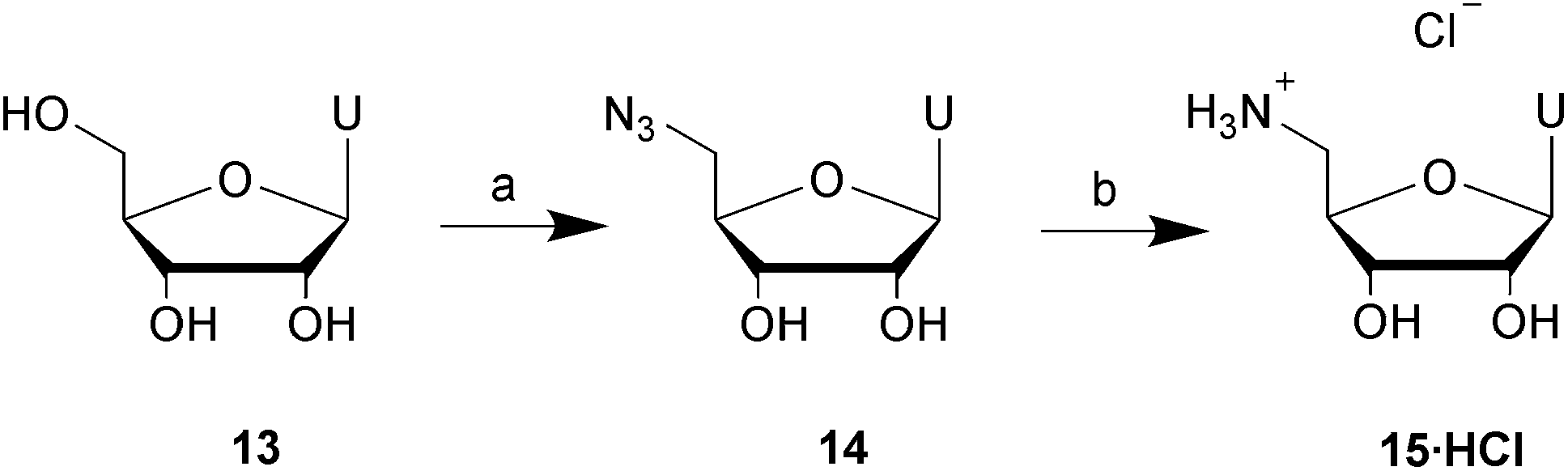 | ||
| Scheme 2 The synthesis of 5′-amino-5′-deoxyuridine, hydrochloride salt 15·HCl. Reagents and conditions: (a) Ph3P, CBr4, LiN3, DMF, 4 h, rt; (b) (i) Ph3P, pyridine, 2 h, rt; (ii) EtOH, HCl(g). | ||
To our knowledge 5′-amino-5′-deoxycytidine 20 has not been reported. We protected the exocyclic amino group of cytosine 16 via benzoylation.28 The protection of the 2′- and 3′-hydroxyl groups and the tosylation of the 5′-hydroxyl group were adapted from procedures developed by Winans and Bertozzi.29 The crude tosylated material was reacted with sodium azide in DMSO in an adapted literature procedure30 to afford azide 18 which was isolated by precipitation in a large volume of water. The protecting groups were removed,31 and 5′-azide 19 was isolated by cation exchange chromatography. The azide was then reduced with triphenylphosphine26 and the amine was isolated as the dihydrochloride salt 20·2HCl (Scheme 3).
 | ||
| Scheme 3 The synthesis of 5′-amino-5′-deoxycytidine, dihydrochloride salt 20·2HCl. Reagents and conditions: (a) (i) benzoic anhydride, MeOH, 5 h, reflux; (ii) 2,2-dimethoxypropane, TsOH, 4 Å mol sieves, DMF, 2 h, 40 °C; (b) (i) Ts2O, pyridine, CH2Cl2, 2.5 h, reflux; (ii) NaN3, DMSO, 2 h 20 min, 70 °C; (c) (i) MeOH, NH4OH, 18 h, rt; (ii) trifluoroacetic acid, H2O, 3 h, rt; (iii) ion exchange chromatography; (d) (i) Ph3P, pyridine, 3 h, rt; (ii) EtOH, HCl(g). | ||
Aminonucleoside phosphorylations and thiophosphorylations
The thiophosphorylations were carried out using both thiophosphoryl chloride 1(S) and thiophosphodichloridate 2(S) on 5′-amino-5′-deoxyadenosine 11, 5′-amino-5′-deoxycytidine 20, 3′-amino-3′-deoxythymidine 12, and 5′-amino-5′-deoxyuridine 15. The oxyphosphorylations were performed using both oxyphosphorylating agents 1(O) and 2(O) on 5′-amino-5′-deoxyadenosine 11 and 3′-amino-3′-deoxythymidine 12. In every case, one equivalent of phosphorylating agent was used, and the phosphorylations were performed at pH 12, based on the findings of our optimisation experiments (Scheme 4).The conversion levels observed in the experiments are shown in Fig. 3. For the thiophosphorylations, conversions were generally high, especially for the reactions of 5′-amino-5′-deoxyguanosine 3 with thiophosphodichloridate 2(S) and 5′-amino-5′-deoxyadenosine 11 with thiophosphoryl chloride 1(S), with conversions of 97% and 99% respectively. A notable exception to the high thiophosphorylation conversions is the reaction with 3′-amino-3′-deoxythymidine 12; this is likely due to the more sterically hindered environment of the 3′-amine nucleophile in this example.
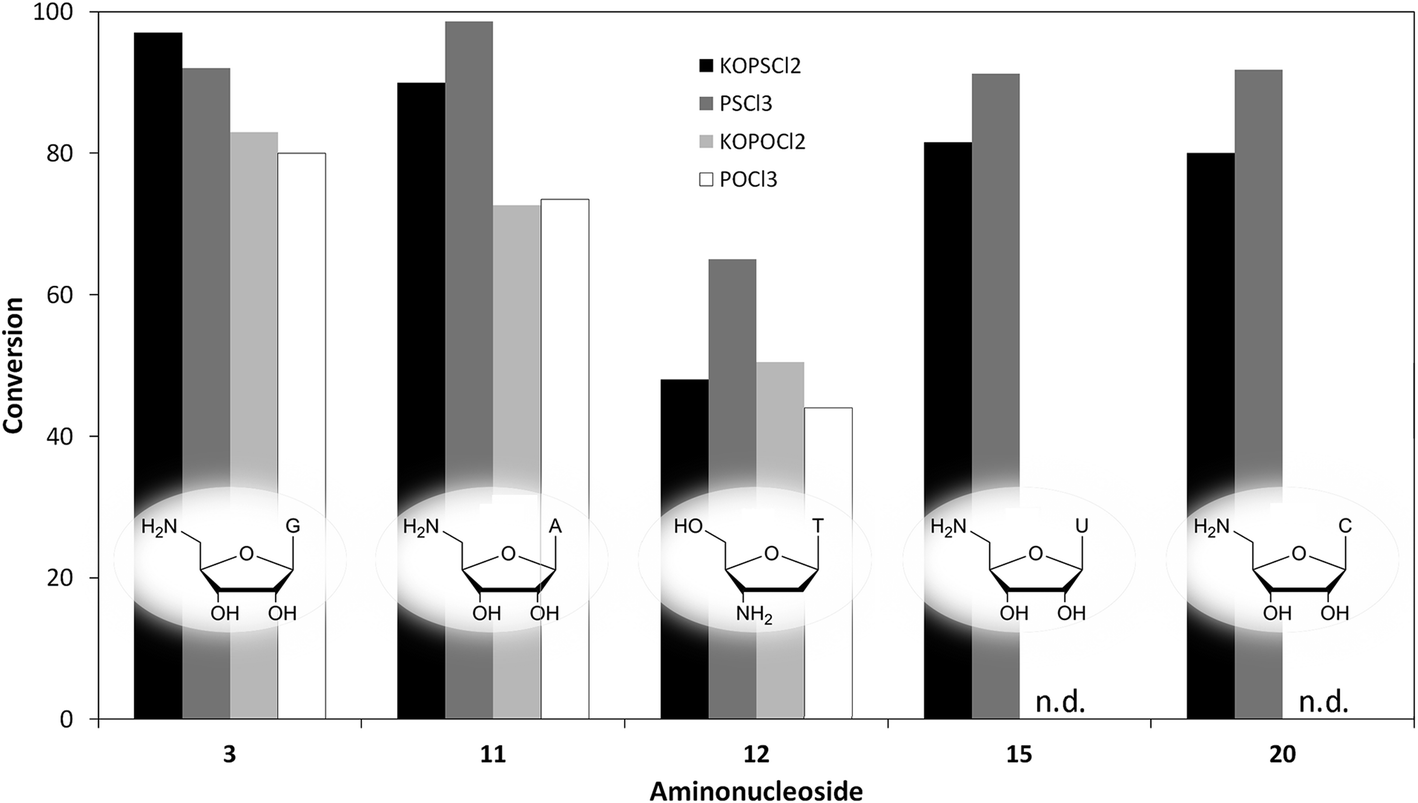 | ||
| Fig. 3 Conversion levels for the N-phosphorylations and N-thiophosphorylations of 5′-amino-5′-deoxyguanosine 3, 5′-amino-5′-deoxyadenosine 11, 3′-amino-3′-deoxythymidine 12, 5′-amino-5′-deoxyuridine 15, and 5′-amino-5′-deoxycytidine 20. | ||
 | ||
| Scheme 4 The N-phosphorylations and N-thiophosphorylations of 5′-amino-5′-deoxyadenosine 11, 3′-amino-3′-deoxythymidine 12, 5′-amino-5′-deoxyuridine 15, and 5′-amino-5′-deoxycytidine 20. | ||
The use of thiophosphoryl chloride 1(S) gave greater levels of conversion than the thiophosphodichloridate ion 2(S), with the exception of the reaction with 5′-amino-5′-deoxyguanosine 3. Unlike the thiophosphodichloridate ion, thiophosphoryl chloride shows very limited solubility in water. This may give rise to the improved selectivity, despite thiophosphoryl chloride's greater reactivity, possibly through reaction at solvent–water interfaces rather than in homogeneous solution.
In other cases, the use of potassium thiophosphodichloridate 2(S) afforded fewer side-products such as the bis thiophosphoramidate 9. For the oxyphosphorylating agents 1(O) and 2(O), the trend appears to be reversed; the phosphodichloridate 2(O) reactions tended to give the greatest levels of conversion, possibly due to the reduced reactivity of 2(O) allowing better mixing and thus greater selectivity than with phosphoryl chloride 1(O). In general, the levels of conversion for the oxyphosphorylations are more modest than those of the thiophosphorylations with a maximum of 83% for the reaction of 5′-amino-5′-deoxyguanosine 3 with phosphodichloridate 2(O). 3′-Amino-3′-deoxythymidine again showed the poorest conversions, at 44% and 51% for the reactions with phosphorylating agents 1(O) and 2(O) respectively.
Thiophosphoramidate S-alkylation
The S-Alkylation of thiophosphoramidates 21a–d(S) could provide access to mimics of naturally occurring phosphodiesters such as CMP-Neu5Ac or analogues for mechanistic studies.4 Additionally, there are many commercially available alkylating agents, and this reaction could allow the rapid development of a diverse range of derivatives. We conducted preliminary studies in this vein recently, where the products of the N-thiophosphorylation-S-alkylation of 5′-amino-5′-deoxyguanosine 3 and 5′-amino-5′-deoxyadenosine 11 were prepared and isolated from non-pH-controlled reactions, and these results served as our starting point.22 Here we investigate the pH optimisation of this procedure. Using our model substrate, 5′-amino-5′-deoxyguanosine 3, we performed N-thiophosphorylation, and the resulting nucleoside-N-thiophosphoramidate anion was then S-alkylated with one of three alkylating agents, each at fixed pHs using an autotitrator (Scheme 5). | ||
| Scheme 5 The alkylation of 5′-deoxyguanosine 5′-thiophosphoramidate 4(S). Reagents and conditions: (a) MeI to give 22a; (b) BnCl to give 22b; or bromoethanol to give 22c. | ||
We chose benzyl chloride, methyl iodide, and 2-bromoethanol as alkylating agents, using guanosine-N-thiophosphoramidate 4(S) as the substrate. Guanosine-N-thiophosphoramidate 4(S) was prepared under optimised conditions, where >95% of the product represented the desired material, and this material was used directly in S-alkylation procedures. The conversion levels determined by 31P NMR spectroscopy for each of the alkylating agents at each pH are illustrated in Fig. 4.
 | ||
| Fig. 4 The conversion levels by 31P NMR spectroscopy of S-alkylations of guanosine-N-thiophosphoramidate 4(S) to S-alkylthiophosphoramidates 22a–c using methyl iodide, benzyl chloride, and 2-bromoethanol, respectively. | ||
To confirm the intermediacy of the unstable, unalkylated precursor N-thiophosphoramidate 4(S), three S-alkylation experiments were performed on 4(S) at pH 12, and the products were isolated by ion exchange chromatography and characterised. Isolated yields of 95%, 63%, and 74% were recorded for the S-methyl 22a, benzyl 22b, and ethan-2-ol 22c systems. S-Alkylation efficiency appeared to be largely unaffected by pH, at least within the range that we studied, with conversions levels being ∼80%. Surprisingly, S-alkylation occurs at lower conversion levels than N-thiophosphorylation, which may be due, in part, to the hydrolysis of the desired products under the high pH conditions. In particular, the formation of a nascent “good” thiolate leaving group through S-alkylation could represent a likely pathway for hydrolysis, and we are currently studying the hydrolysis kinetics of the S-alkylated thiophosphoramidates to give greater insight.
Conclusions
Effective N-oxyphosphorylation and N-thiophosphorylation of aminonucleosides 3, 11, 12, 15 and 20 was achieved through pH control, with high levels of conversion. Phosphodichloridates 2(O) and 2(S) were competent alternative phosphorylation agents, that eliminated the formation of undesired bis-amino(thio)phosphoramidates, while retaining similar levels of conversion. The S-alkylation of guanosine-N-thiophosphoramidate 4(S) showed no significant pH-sensitivity across the pH range 9–12, although conversion levels were not quantitative. The combination of efficient N-(thio)phosphorylation and S-alkylation gives straightforward access to phosphodiester mimics from a range of unprotected aminonucleosides derived from the common nucleobases.Experimental
N4-Benzoylcytidine
In an adapted literature procedure,28 cytidine (2.784 g, 11.4 mmol) and benzoic anhydride (2.818 g, 12.5 mmol) were placed in a round bottomed flask with dry methanol (300 ml) and heated at reflux with stirring. After 1 h, additional benzoic anhydride (2.774 g, 12.3 mmol) was added, followed by further additions (2.785 g, 12.3 mmol after 2 h, 2.794 g, 12.4 mmol after 3 h). Heating was maintained for a total of 5 h. After allowing the reaction vessel to cool for 18 h, the precipitate was collected by vacuum filtration. Drying over P2O5 under vacuum yielded the desired product (2.786 g, 70%), mp 236–238 °C (from methanol) (lit.,32 238–240 °C); νmax/cm−1 3421, 3307, 3161, 1644 (CO); δH (400 MHz, (CD3)2SO) 3.61 (1H, ddd, J 12.3, 5.2, 3.1, C5′-Hα), 3.76 (1H, ddd, J 12.3, 5.2, 2.8, C5′-Hβ), 3.92 (1H, td, J 5.9, 2.9, C4′-H), 3.95–4.05 (2H, m, C2′-H, C3′-H), 5.05 (1H, d, J 5.7, C3′-OH), 5.18 (1H, app t, J 5.1, C5′-OH), 5.51 (1H, d, J 4.8, C2′-OH), 5.81 (1H, d, J 2.9, C1′-H), 7.34 (1H, d, J 7.5, C5-H), 7.52 (2H, dd, J 7.1, 8.4, m-Ph), 7.63 (1H, t, J 7.4, p-Ph), 8.00 (2H, d, J 7.5, o-Ph), 8.51 (1H, d, J 7.5, C6-H), 11.25 (1H, s, NH); δC (126 MHz, (CD3)2SO) 60.6, 69.3, 75.3, 84.9, 90.9, 96.6, 110.0, 129.2, 133.5, 133.8, 146.1, 155.4, 163.8, 168.0; m/z 346.2 (M − H, 100%) 347.2 (26) 693 (22).N4-Benzoyl-2′,3′-O-isopropylidenecytidine (17)
In an adapted literature procedure,29 N4-benzoylcytidine (2.79 g, 8.03 mmol), 2,2-dimethoxypropane (11 ml), 4 Å molecular sieves, and tosic acid monohydrate (0.45 g, 2.37 mmol) were placed in a round-bottomed flask with DMF (44 ml). The mixture was heated at 40 °C for 2.5 h, before Amberlyst® A-21 anion exchanger (1.19 g) was added and heating was continued for a further 30 min. The mixture was filtered through Celite®, the solvent was then removed under vacuum from the filtrate, and the residue was recrystallised from water to yield the product (2.375 g, 76%); mp 163–165 °C; νmax/cm−1 1652 (CO), 1482, 1303; δH (400 MHz, CDCl3) 1.37 (3H, s, CH3α), 1.63 (3H, s, CH3β), 3.54 (1H, br s, C5′-OH), 3.80–3.89 (1H, m, C5′-Hα), 3.97 (1H, dd, J 12.2, 2.3, C5′-Hβ), 4.40 (1H, app q, J 3.0, C4′-H), 5.09 (1H, dd, J 6.4, 3.3, C3′-H), 5.25 (1H, dd, J 6.6, 2.6, C2′-H), 5.55 (1H, d, J 2.7, C1′-H), 7.52 (2H, d, J 7.8, m-Ph), 7.63 (1H, t, J 7.4, p-Ph), 7.81 (1H, d, J 7.4, C6-H), 7.89 (2H, d, J 7.5, o-Ph), 8.77 (1H, s, NH); δC (126 MHz, CDCl3) 25.2, 27.3, 62.9, 80.5, 83.6, 88.1, 99.0, 100.0, 112.8, 114.1, 127.3, 127.6, 128.6, 129.1, 133.4, 143.3, 166.1; m/z 388.1500 (M + H+, 20%), requires 388.1509, 410.1325 (M + Na+, 100).N4-Benzoyl-2′,3′-O-isopropylidene-5′-O-toluenesulfonylcytidine
N4-Benzoyl-2′,3′-O-isopropylidenecytidine (2.10 g, 5.42 mmol), 4-toluenesulfonyl chloride (2.44 g, 12.9 mmol), pyridine (20 ml) and DCM (40 ml) were placed in a round-bottomed flask and heated at reflux for 2.5 h. The solution was then diluted with chloroform (120 ml) and washed with hydrochloric acid (0.5 M, 5 × 50 ml) and saturated sodium hydrogencarbonate solution (2 × 50 ml). The organic layer was dried over MgSO4 and the solvent was removed under vacuum to yield the crude product, which was used in the next reaction without further purification. (1.53 g).5′-Azido-5′-deoxy-N4-benzoyl-2′,3′-O-isopropylidenecytidine (18)
Adapting a literature procedure,30 N4-benzoyl-2′,3′-O-isopropylidene-5′-O-toluenesulfonylcytidine (1.08 g, 1.99 mmol) and sodium azide (579 mg, 8.91 mmol) were placed in a flask with DMSO (10 ml) and heated at 70 °C for 2 h 20 min. The solution was then added to H2O (550 ml) and left to stir for 18 h. The precipitated product was then isolated by vacuum filtration. (430 mg, 52%); mp 163–165 °C; νmax/cm−1 3354, 1640 (CO), 1485; δH (400 MHz, CDCl3) 1.37 (3H, s, CH3α), 1.60 (3H, s, CH3β), 3.65 (1H, dd, J 12.8, 4.4, C5′-Hα), 3.76, (1H, dd, J 12.8, 6.8, C5′-Hβ) 4.31–4.36 (1H, m, C4′-H), 4.90 (1H, dd, J 6.4, 4.0, C2′-H), 5.11–5.15 (1H, m, C3′-H), 5.70 (1H, d, J 1.5, C1′-H), 7.53 (2H, t, J 7.6, m-Ph), 7.63 (1H, t, J 7.4, p-Ph), 7.77 (1H, d, J 7.4, C6-H), 7.89 (2H, d, J 7.2, o-Ph), 8.67 (1H, s, NH); δC (100 MHz, D2O) 25.2, 27.3, 62.8, 77.2, 80.5, 83.8, 88.1, 97.0, 98.7, 114.1, 127.7, 129.1, 132.7, 133.4, 147.8, 163.1; m/z 413.1585 (M + H+, 100%), requires 413.1573, 435.1406 (M + Na+, 40%).5′-Azido-5′-deoxycytidine (19)
Based on a literature procedure,31 5′-Azido-5′-deoxy-N4-benzoyl-2′,3′-O-isopropylidene cytidine (732 mg, 1.78 mmol) was placed in a round-bottomed flask with a 1:1 mixture of methanol and concentrated ammonium hydroxide (90 ml) and was stirred at room temperature for 18 h. The solvents were removed under vacuum, and the residue was stirred in a 9:1 mixture of trifluoroacetic acid and H2O for 3 h at room temperature before the solvents were removed under vacuum. The residue was dissolved in H2O and introduced to a protonated SP Sepharose® Fast Flow cation exchange column (1.6 cm i.d. × 22 cm, 5 ml min−1 flow rate). The nucleoside was eluted with a 3% ammonium hydroxide solution (diluted from 35% (w/w) ammonium hydroxide solution) and the eluted fractions were freeze-dried. The residue was then dissolved in methanol and filtered, and the solvent was removed from the filtrate under vacuum to give the solid product. (334 mg, 70%); νmax/cm−1 2104 (N3), 1645 (CO), 1438; δH (400 MHz, D2O) 3.64 (1H, dd, J 13.7, 5.0, C5′-Hα), 3.78 (1H, dd, J 13.7, 3.1, C5′-Hβ), 4.14–4.19 (1H, m, C4′-H), 4.21 (1H, dd, J 6.2, 5.3, C3′-H), 4.34 (1H, dd, J 5.3, 4.0, C2′-H), 5.87 (1H, d, J 4.0, C1′-H), 6.03 (1H, d, J 7.5, C5-H), 7.74 (1H, d, J 7.6, C6-H); δC (100 MHz, D2O) 51.2, 69.9, 73.3, 81.5, 90.5, 96.3, 141.6, 157.4, 163.8; m/z 267.2 (M − H, 100%) 268.3 (15).
5′-Amino-5′-deoxycytidine, hydrochloride salt (20·2HCl)
5′-Azido-5′-deoxycytidine (400 mg, 1.49 mmol) was placed in a flask with triphenylphosphine (790 mg, 3.01 mmol) and pyridine (6 ml). The solution was stirred at room temperature for 3 h before further triphenylphosphine (791 mg, 3.02) and pyridine (6 ml) were added. The solution was stirred for an additional 3 h at room temperature before ammonia solution (50 ml, 35% w/w) was added, whereupon immediate precipitation was observed. The mixture was stirred overnight before being diluted with water (100 ml) and extracted with chloroform (3 × 100 ml). Residual chloroform was removed from the aqueous layer under vacuum, and the aqueous extracts were lyophilised. The resulting solid was dissolved in ethanol (60 ml) and hydrogen chloride gas was bubbled through the solution. The addition of diethyl ether (300 ml) precipitated the product, which was isolated by filtration, washed with a small quantity of diethyl ether, and dried under vacuum overnight (382 mg, 81%); mp 190–191 °C (decomp.) (from ethanol); νmax/cm−1 3118, 3046, 1715, 1673, 1404; δH (400 MHz, D2O) 3.34 (1H, dd, J 13.6, 9.4, C5′-Hα), 3.47 (1H, dd, J 13.6, 3.0, C5′-Hβ), 4.18–4.31 (2H, m, C3′-H, C4′-H), 4.51 (1H, dd, J 3.4, 5.5, C2′-H), 5.77 (1H, d, J 3.4, C1′-H), 6.24 (1H, d, J 7.9, C5-H), 7.87 (1H, d, J 7.9, C6-H); δC (100 MHz, D2O) 41.9, 71.4, 73.5, 80.2, 94.1, 95.8, 137.8, 145.9, 160.9; m/z 243.1095 (M + H+, 50%), requires 243.1093, 265.0907 (M + Na+, 100).3′-Amino-3′-deoxythymidine, hydrochloride salt (12·HCl)
Adapting a literature procedure,26 3′-amino-3′-deoxythymidine (1.00 g, 3.74 mmol) and triphenylphosphine (1.54 g, 5.87 mmol) were dissolved in pyridine (8 ml) and stirred at room temperature for 1 h. Ammonia solution (30 ml, 35%) was then added, and the mixture left to stir overnight. The suspension was diluted with water (30 ml) and extracted with chloroform (3 × 30 ml) before being lyophilised. The solid residue was dissolved in ethanol (100 ml) and hydrogen chloride gas was bubbled through the solution until precipitation was observed. The precipitate was isolated by filtration, and washed with a small quantity of diethyl ether. Additional product was obtained by adding diethyl ether (500 ml) to the filtrate, and again filtering and washing the precipitate. The isolated solids were combined and dried under vacuum overnight yielding a total of 846 mg, 81%; mp 253–255 °C (decomp.) (from ethanol); νmax/cm−1 3392, 3032, 1694, 1644, 1470. δH (400 MHz, D2O) 1.86 (3H, s, C5-CH3), 2.54–2.70 (2H, m, C2′-HaHb), 3.81 (1H, dd, J 12.6, 4.6, C5′-Hα), 3.89 (1H, dd, J 12.6, 3.4, C5′-Hβ), 4.06 (1H, dt, J 8.1, 5.5, C3′-H), 4.19–4.28 (1H, m, C4′-H), 6.25–6.32 (1H, t, J 6.8, C1′-H), 7.62 (1H, d, J 1.1, C6-H); δC (100 MHz, D2O) 10.5, 33.8, 49.1, 59.7, 81.5, 84.1, 110.5, 136.6, 150.5, 165.4; m/z 242.1142 (M + H+, 100%), requires 242.1141, 264.0961 (M + Na+, 90).Lithium azide
Following a literature procedure,33 sodium azide (6.52 g, 100 mmol) and lithium sulfate (6.90 g, 62.8 mmol) were co-dissolved in water (35 ml) and stirred for 10 min. Ethanol (175 ml) was slowly added, and the mixture was stirred for a further 10 min. The precipitate was removed by filtration, and the solvent was removed from the filtrate under vacuum. The solid residue was dried on the high vacuum line to yield the product (4.818 g, 98%).5′-Azido-5′-deoxyuridine (14)
With some modifications, a literature procedure was followed.27 Uridine (5.00 g, 20.5 mmol), triphenylphosphine (8.05 g, 30.5 mmol), tetrabromomethane (10.2 g, 30.5 mmol), and lithium azide (3.695 g, 75.5 mmol) were stirred together in DMF (100 ml) for 4 h at room temperature. The greater part of the solvent was removed under vacuum, and the residue was extracted with chloroform (100 ml) and water (100 ml), with the aqueous layer being retained. The aqueous layer was washed again with chloroform (2 × 100 ml) before being lyophilised. The viscous residue was purified by column chromatography, using an isocratic 5:1 mixture of chloroform and methanol. The resulting oil was dissolved in 5:1 chloroform–methanol and adsorbed on to a plug of silica, which was washed with 100% chloroform to remove residual DMF, then released using the 5:1 chloroform–methanol solution to yield the purified product. (3.70 g, 67%); νmax/cm−1 2102 (N3), 1667 (CO), 1462, 1386, 1100 (R2COH); δH (500 MHz, D2O) 3.69 (1H, dd, J 13.7, 5.0, C5′-Hα), 3.82 (1H, dd, J 13.7, 3.2, C5′-Hβ), 4.18–4.23 (1H, m, C4′-H), 4.27 (1H, app t, J 5.6, C′3-H), 4.42 (1H, dd, J 5.4, 4.5, C2′-H), 5.89 (1H, d, J 4.4, C1′-H), 5.93 (1H, d, J 8.1, C5-H), 7.80 (1H, d, J 8.1, C6-H); δC (126 MHz, D2O) 51.6, 70.2, 73.3, 82.3, 90.1, 102.6, 142.2, 151.7, 166.2; m/z 268.0676 (M − H, 30%), requires 268.0682, 304.0408 (100).
5′-Amino-5′-deoxyuridine, hydrochloride salt (15·HCl)
Adapting a literature procedure,26 5′-azido-5′-deoxyuridine (404 mg, 1.50 mmol) and triphenylphosphine were (1.66 g, 6.33 mmol) were dissolved in pyridine (1.6 ml) and stirred for 2 h. Ammonia solution (35%, 12 ml) was then added, and the mixture left to stir overnight. Water (40 ml) was added, and the mixture was extracted with chloroform (3 × 40 ml). Residual chloroform was then removed from the aqueous layer under vacuum, and the remaining solution was lyophilised. The resulting powder was then dissolved in ethanol (20 ml) and heated at reflux until dissolved. Hydrogen chloride gas was bubbled through the solution until precipitation occurred, and the pure hydrochloride salt was isolated by filtration and washing with a small quantity of ethanol. (329 mg, 78%); mp 210–215 °C (decomp.) (from ethanol); νmax/cm−1 3040, 1666 (CO), 1464, 1042; δH (700 MHz, D2O) 3.29–3.36 (1H, m, C5′-Hα), 3.44–3.49 (1H, m, C5′-Hβ), 4.22–4.26 (2H, m, C′3-H, C4′-H), 4.52 (1H, dd, J 5.2, 4.0, C2′-H), 5.77 (1H, d, J 4.0, C1′-H), 5.89 (1H, d, J 8.0, C5-H), 7.66 (1H, d, J 8.1, C6-H); δC (126 MHz, D2O) 41.8, 71.4, 73.2, 80.0, 93.3, 102.8, 143.9, 152.0, 166.9; m/z 244.0934 (M − H, 100%), requires 244.0933.Phosphorylating agents
The preparation of potassium phosphodichloridate and potassium thiophosphodichloridate has been reported previously.20,34Phosphorylation procedure
The aminonucleoside (0.500 mmol) was placed in a thermostated reaction flask maintained at 25 °C and water and potassium hydroxide were added as required to make a 5 ml solution at the desired pH. The phosphorylating agent (1.50 ml, 0.333 M in MeCN) was added using a Hamilton® microlitre syringe. The addition took place over 10 min with vigorous stirring, and with the tip of the syringe below the surface of the reaction mixture. Throughout the experiment, the pH was kept constant using a 1 M solution of potassium hydroxide, added by the autotitrator system. The experiment was considered to be complete when the autotitrator needed to add negligible quantities of potassium hydroxide solution to the reaction mixture. After the completion of the reaction, the organic solvent was removed under vacuum and aqueous remainder was lyophilised.Alkylation procedure
Aqueous solutions (5 ml) of 5′-amino-5′-deoxyguanosine thiophosphoramidate 114 (28.2 mg) were made up in the thermostated reaction flask at a concentration of 20 mM at the desired pH, and were vigorously stirred at 25 °C. Methyl iodide (14.3 μl), benzyl chloride (26.5 μl) or bromoethanol (12.4 μl) alkylating agent was then added to the reaction solution via Hamilton® syringe and stirred for 1 h. The excess alkylating agents were removed by extraction with diethyl ether (3 × 10 ml) and the aqueous solution was subsequently lyophilised. The conversions of 5′-amino-5′-deoxyguanosine thiophosphoramidate to alkylated thiophosphoramidate product were estimated by 31P NMR spectroscopy. Where the products of the alkylation were isolated, the lyophilised crude material was redissolved in water (4 ml) and purified by anion exchange chromatography with a flow rate of 5 ml min−1 over a DEAE-Sepharose® resin. Triethylammonium bicarbonate buffer was applied in a 0 to 0.15 M gradient over 200 min. The fractions corresponding to peaks in the UV-vis absorbance were collected and lyophilised.5′-Amino-5′-deoxyguanosine, N-,S-methylthiophosphoramidate, triethylammonium salt (22a)
After purification, a small quantity of by-product (MeSPO32−, <10% mol basis by 31P NMR spectroscopy) remained. 47 mg, 95%; δH (400 MHz, D2O) 1.26 (9H, t, J 7.3, HN+(CH2CH3)3), 2.08 (3H, d, J 13.0, CH3S), 2.94–3.28 (2H, m, C5′-H2), 3.18 (6H, q, J 7.3, HN+(CH2CH3)3), 4.31–4.35 (1H, m, C4′-H), 4.41 (1H, app d, J 5.6, C3′-H), 5.08 (1H, dd, J 8.0, 5.6, C2′-H), 5.76 (1H, d, J 8.0, C1′-H), 7.87 (1H, s, C8-H); δC (101 MHz, D2O) 8.3, 11.4, 43.4, 46.7, 71.4, 86.0, 88.8, 100.0, 117.2, 139.8, 151.3, 153.7, 159.0; δP (162 MHz, D2O) 26.3; m/z 391.0597 (M − Et3NH+, 100%), requires 391.0590, 392.0627 (20), 393.0617 (10).5′-Amino-5′-deoxyguanosine, N-,S-benzylthiophosphoramidate, triethylammonium salt (22b)
After the purification procedure, some by-product (BnSPO32−, ∼10% mol basis by 31P NMR spectroscopy) remained. 36 mg, 63%; δH (400 MHz, D2O) 1.25 (9H, t, J 7.3, HN+(CH2CH3)3), 2.80–3.20 (2H, m, C5′-H2), 3.17 (6H, q, J 7.3, HN+(CH2CH3)3), 3.80–3.88 (2H, m, J 5.6, SCH2Ph), 4.18–4.22 (1H, m, C4′-H), 4.33 (1H, dd, J 5.6, 1.6, C3′-H), 5.10 (1H, dd, J 7.9, 5.6, C2′-H), 5.66 (1H, d, J 7.9, C1′-H) 6.99 (1H, t, J 7.1, o-ph), 7.06 (2H, t, J 7.1, m-ph), 7.24 (2H, d, J 7.1, o-ph) 7.82 (1H, s, C8-H); δC (101 MHz, D2O) 8.2, 10.5, 43.3, 46.6, 59.0, 70.9, 71.5, 85.7, 88.7, 117.2, 126.6, 128.2, 138.6, 139.9, 151.3, 153.5, 158.9; δP (162 MHz, D2O) 24.1; m/z 467.0899 (M − Et3NH+, 100%), requires 467.0903, 468.0936 (20), 469.0919 (10).5′-Amino-5′-deoxyguanosine, N-,S-ethan-2-olthiophosphoramidate, triethylammonium salt (22c)
39 mg, 74%; δH (400 MHz, D2O) 1.35 (9H, t, J 7.3, HN+(CH2CH3)3), 2.88 (2H, dt, J 12.7, 6.4, SCH2CH2OD), 3.26 (6H, q, J 7.3, HN+(CH2CH3)3), 3.79 (2H, t, J 6.4, SCH2CH2OD), 4.43 (1H, dd, J 2.9, 1.6, C4′-H), 4.50 (1H, dd, J 5.6, 1.6, C3′-H), 5.18 (1H, dd, J 8.0, 5.6, C2′-H) 5.86 (1H, d, J 8.0, C1′-H), 7.95 (1H, s, C8-H); δC (101 MHz, D2O) 8.3, 10.6, 31.9, 43.4, 46.7, 61.5, 71.4, 85.9, 88.7, 117.4, 139.4, 151.3, 155.0, 160.5; δP (162 MHz, D2O) 24.9; m/z 421.0679 (M − Et3NH+, 100%), requires 421.0695, 422.0733 (20), 443.0501 (10).Analysis
The solid residues resulting from the experiments were dissolved in 750 μl or the minimum volume of D2O, whichever was less, and analysed by NMR spectroscopy to determine conversion. The conversion levels were determined using all the peaks visible by 31P NMR spectroscopy with a relaxation time of 10 s, acquisition time of 1.28 s, and 128 transients. 1H NMR spectra were also recorded. The 31P NMR spectroscopic chemical shifts of the phosphoramidate and thiophosphoramidate products of the reaction were found to be consistent with those determined in our previous work.15,19,22 The changes in chemical shifts of the 1H NMR signals corresponding to the 5′ or 3′ protons (depending on the substrate) of ∼0.5 ppm upfield, and the increase in multiplicity through coupling to phosphorus corroborated the formation of the desired phosphoramidates and thiophosphoramidates.Acknowledgements
This work was supported by EPSRC DTA funding (LPC and RJD), the Royal Society and Durham University.Notes and references
- S. Spitzer and F. Eckstein, Nucleic Acids Res., 1988, 16, 11691–11704 CrossRef CAS PubMed.
- J. K. Chen, R. G. Schultz, D. H. Lloyd and S. M. Gryaznov, Nucleic Acids Res., 1995, 23, 2661–2668 CrossRef CAS PubMed.
- A. Grandas, W. S. Marshall, J. Nielsen and M. H. Caruthers, Tetrahedron Lett., 1989, 30, 543–546 CrossRef CAS.
- H. J. Korhonen, L. P. Conway and D. R. W. Hodgson, Curr. Opin. Chem. Biol., 2014, 21, 63–72 CrossRef CAS PubMed.
- A. Hampton, Biochim. Biophys. Acta, 1970, 198, 594–600 CrossRef CAS.
- A. Hampton, L. W. Brox and M. Bayer, Biochemistry, 1969, 8, 2303–2311 CrossRef CAS.
- A. B. Burgin and N. R. Pace, EMBO J., 1990, 9, 4111–4118 CAS.
- G. Sengle, A. Jenne, P. S. Arora, B. Seelig, J. S. Nowick, A. Jaschke and M. Famulok, Bioorg. Med. Chem., 2000, 8, 1317–1329 CrossRef CAS.
- M. Skipsey, G. Hack, T. A. Hooper, M. C. Shankey, L. P. Conway, M. Schröder and D. R. W. Hodgson, Nucleosides, Nucleotides Nucleic Acids, 2013, 32, 670–681 CAS.
- E. Paredes, M. Evans and S. R. Das, Methods, 2011, 54, 251–259 CrossRef CAS PubMed.
- V. J. Davisson, D. R. Davis, V. M. Dixit and C. D. Poulter, J. Org. Chem., 1987, 52, 1794–1801 CrossRef CAS.
- M. Yoshikawa, T. Kato and T. Takenishi, Bull. Chem. Soc. Jpn., 1969, 42, 3505–3508 CrossRef CAS.
- J. Nielsen, M. Taagaard, J. E. Marugg, J. H. Vanboom and O. Dahl, Nucleic Acids Res., 1986, 14, 7391–7403 CrossRef CAS PubMed.
- D. Williamson and D. R. W. Hodgson, Org. Biomol. Chem., 2008, 6, 1056–1062 CAS.
- D. Williamson, M. J. Cann and D. R. W. Hodgson, Chem. Commun., 2007, 5096–5098 RSC.
- M. Trmcic and D. R. W. Hodgson, Chem. Commun., 2011, 47, 6156–6158 RSC.
- K. Schattka and B. Jastorff, Chem. Ber./Recl., 1974, 107, 3043–3052 CrossRef CAS PubMed.
- B. Jastorff and H. Hettler, Chem. Ber./Recl., 1969, 102, 4119–4127 CrossRef CAS PubMed.
- M. Trmčić and D. R. W. Hodgson, Chem. Commun., 2011, 47, 6156–6158 RSC.
- R. J. Delley, A. C. Donoghue and D. R. W. Hodgson, J. Org. Chem., 2012, 77, 5829–5831 CrossRef CAS PubMed.
- D. K. Dean, Synth. Commun., 2002, 32, 1517–1521 CrossRef CAS PubMed.
- M. Trmcic, F. L. Chadbourne, P. M. Brear, P. W. Denny, S. L. Cobb and D. R. W. Hodgson, Org. Biomol. Chem., 2013, 11, 2660–2675 CAS.
- S. J. Benkovic and E. J. Sampson, J. Am. Chem. Soc., 1971, 93, 4009–4016 CrossRef CAS.
- Nucleic Acids in Chemistry and Biology, ed. G. M. Blackburn and M. J. Gait, Oxford University Press, New York, 2 edn, 1996 Search PubMed.
- M. Kolb, C. Danzin, J. Barth and N. Claverie, J. Med. Chem., 1982, 25, 550–556 CrossRef CAS.
- P. Cano-Soldado, M. Molina-Arcas, B. Alguero, I. Larrayoz, M. P. Lostao, A. Grandas, F. J. Casado and M. Pastor-Anglada, Biochem. Pharmacol., 2008, 75, 639–648 CrossRef CAS PubMed.
- I. Yamamoto, M. Sekine and T. Hata, J. Chem. Soc., Perkin Trans. 1, 1980, 306–310 RSC.
- K. A. Watanabe and J. J. Fox, Angew. Chem., Int. Ed., 1966, 5, 579–580 CrossRef CAS PubMed.
- K. A. Winans and C. R. Bertozzi, Chem. Biol., 2002, 9, 113–129 CrossRef CAS.
- M. G. Stout, M. J. Robins, R. K. Olsen and R. K. Robins, J. Med. Chem., 1969, 12, 658–661 CrossRef CAS.
- G. H. Jones, M. Taniguchi, D. Tegg and J. G. Moffatt, J. Org. Chem., 1979, 44, 1309–1317 CrossRef CAS.
- P. S. Ludwig, R. A. Schwendener and H. Schott, Synthesis, 2002, 2387–2392 CrossRef CAS PubMed.
- R. Steudel and P. W. Schenk, in Handbuch der Präparativen Anorganischen Chemie, ed. G. Brauer , Ferdinand Enke Verlag, Stuttgart, 1975, 1, p. 475 Search PubMed.
- Y. Segall, J. Agric. Food Chem., 2011, 59, 2845–2856 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra. See DOI: 10.1039/c4ra08317b |
| This journal is © The Royal Society of Chemistry 2014 |