
NHC-catalyzed one-pot oxidation and oxidative esterification of allylic alcohols using TEMPO: the effect of alcohol additives†
Ye-Won Kanga and
Hye-Young Jang*ab
aDepartment of Energy Systems Research, Ajou University, Suwon 443-749, Korea. E-mail: hyjang2@ajou.ac.kr; Fax: +82 31 2191615; Tel: +82 31 219 2555
bKorea Carbon Capture & Sequestration R&D Center, Deajeon 305-343, Korea
First published on 9th September 2014
Abstract
A combination of N-heterocyclic carbene (NHC) catalysts and 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) was proposed for the synthesis of allylic esters from allylic alcohols. The yield of one-pot conversion of allylic alcohols to esters increased when hexafluoroisopropanol (HFIP) was used. The effect of HFIP in this tandem reaction was investigated by monitoring the reaction using gas chromatographic analysis. Control experiments using oxoammonium showed that the oxidative esterification occurred via a single-electron transfer mechanism.
Powerful and selective radicals such as 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) and related nitroxides have been used in diverse oxidation reactions.1–3 For example, the oxidation of alcohols, oxidative bond-forming reactions, and rearrangement reactions occur through nitroxide radicals. Recently, our research group published N-heterocyclic carbene (NHC)-catalyzed oxidative esterification, thioesterification, and amidation of aldehydes using TEMPO, inspired by Studer's carbene-catalyzed TEMPO-ester formation.4
In the previous publication, we reported the one-pot conversion of allylic alcohols to esters with modest yields in the presence of carbene catalysts and 3 equiv. of TEMPO.4b As an extension of such NHC-catalyzed TEMPO-mediated oxidative reactions, we present herein a one-pot reaction of the oxidation of allylic alcohols and subsequent esterification of aldehydes in the presence of reduced amounts of carbene catalysts and TEMPO.4b,4c TEMPO radicals were recycled in the absence of chemical oxidants other than air. The improved results including the higher yields with 1 equiv. and less than 1 equiv. of TEMPO were explained by gas chromatographic (GC) analysis and control experiments. In addition, although most of the TEMPO-mediated oxidation reactions are known to proceed via two-electron oxidation by oxoammonium,3 we present that our NHC-catalyzed oxidative reactions using TEMPO undergo single-electron transfer (SET) oxidation.4–6 Although there are abundant examples of transition-metal-catalyzed oxidation of alcohols to esters, a metal-free carbene-catalyzed one-pot oxidation of alcohols to esters has not been actively reported.4b,7,8 Therefore, our metal-free TEMPO-mediated conversion of allylic alcohols to esters provides useful synthetic and mechanistic information for further development of TEMPO-mediated oxidative reactions.
The optimization results of one-pot oxidation/oxidative esterification of cinnamyl alcohol 1a are listed in Table 1. We have previously reported one-pot oxidation/oxidative esterification of allylic alcohols in the presence of 1,3-bis-(2,4,6-trimethylphenyl)imidazol-2-ylidene (IMes) and TEMPO (entries 1 and 2).4b In the absence of alcohol additives, 1a was converted to 1b in 45% yield by using IMes (10 mol%) and TEMPO (2 equiv.). The yield increased to 59% upon adding 0.2 equiv. of ethanol. By changing the alcohol to HFIP, in this study, the yield was increased to 84% (entry 3). Other halogenated ethanol derivatives (CCl3CH2OH and CF3CH2OH) showed results similar to those obtained using ethanol (entries 4 and 5). The addition of phenol enhanced the yield of 1b, which was yet lower than that of the reaction with HFIP (entry 6). The reaction with a more acidic phenol derivative, pentafluorophenol, provided 1b in a lower yield (entry 7). A different carbene catalyst, 1,3-bis-(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr), was tested to obtain 1b in 69% yield (entry 8). With lower IMes loadings (5 mol%), the yield of 1b slightly decreased to 78% (entry 9). However, by varying the amount of TEMPO (1 equiv., 0.75 equiv., and 0.5 equiv.), it was found that the yield was retained to above 86% upon the addition of 1 equiv. and 0.75 equiv. of TEMPO but not for 0.5 equiv. (entries 10–12). It was also found that in the absence of HFIP the yield was decreased to 12% (entry 13). When HFIP was used as a solvent, the product was not formed. Thus, 0.2 equiv. of HFIP was added to the reaction mixture to improve the yield of 1b.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | TEMPO | Additive (equiv.) | Yield |
| 1 | IMes (10 mol%) | 2 equiv. | — | 45% |
| 2 | IMes (10 mol%) | 2 equiv. | EtOH (0.2) | 59% |
| 3 | IMes (10 mol%) | 2 equiv. | HFIP (0.2) | 84% |
| 4 | IMes (10 mol%) | 2 equiv. | CCI3CH2OH (0.2) | 58% |
| 5 | IMes (10 mol%) | 2 equiv. | CF3CH2OH (0.2) | 54% |
| 6 | IMes (10 mol%) | 2 equiv. | PhOH (0.2) | 70% |
| 7 | IMes (10 mol%) | 2 equiv. | F5-PhOH (0.2) | 62% |
| 8 | IPr (10 mol%) | 2 equiv. | HFIP (0.2) | 69% |
| 9 | IMes (5 mol%) | 2 equiv. | HFIP (0.2) | 78% |
| 10 | IMes (5 mol%) | 1 equiv. | HFIP (0.2) | 89% |
| 11 | IMes (5 mol%) | 0.75 equiv. | HFIP (0.2) | 86% |
| 12 | IMes (5 mol%) | 0.5 equiv. | HFIP (0.2) | 59% |
| 13 | IMes (5 mol%) | 1 equiv. | — | 12% |
 |
||||

In the reaction shown in Table 1, 1a underwent two-step oxidation, prior to the reaction with another equivalent of 1a, which acted as an alcohol in the esterification reaction. Theoretically, 2 equiv. of TEMPO should be consumed because half of the amount of 1a underwent alcohol oxidation followed by oxidative esterification of the aldehydes, which required two-electrons in each step. As shown in Table 1, less than 1 equiv. of TEMPO appears to be sufficient for the two oxidation processes in the presence of HFIP. To determine the role of HFIP, alcohol oxidation and oxidative esterification were analyzed separately. First, alcohol oxidation was conducted using p-OMe-substituted cinnamyl alcohol 2a in the absence of carbene catalysts (Table 2). Without HFIP, the reaction using 1 equiv. of TEMPO afforded 2c in 66% yield (entry 1). By adding 0.2 equiv. of HFIP and 1 equiv. of TEMPO, the yield was enhanced to 78% (entry 2). Keeping the amount of HFIP constant and reducing the amount of TEMPO to 0.5 equiv. resulted in 34% yield (entry 3). The oxidation of 2a was slightly improved by HFIP.
|
|||
|---|---|---|---|
| Entry | TEMPO | HFIP | Yield |
| 1 | 1 equiv. | — | 66% |
| 2 | 1 equiv. | 0.2 equiv. | 78% |
| 3 | 0.5 equiv. | 0.2 equiv. | 34% |

Next, the effect of HFIP in oxidative esterification was analyzed (Table 3). Under the specified reaction conditions, benzyl alcohol was not oxidized by TEMPO to yield benzaldehyde, implying that in this reaction TEMPO was used only for the oxidative esterification of 1c. In the absence of HFIP, oxidative esterification product 1d was formed in 37% yield (entry 1). Upon the addition of HFIP, the yield of 1d increased to 66% (entry 2). Even with only 0.5 equiv. of TEMPO, the yield of 1d was 41%, which was higher than the result of entry 1 (entry 3). A comparison of the results of Tables 2 and 3 showed that the effect of HFIP is probably more critical in the oxidative esterification than in the oxidation of alcohols.
|
|||
|---|---|---|---|
| Entry | TEMPO | HFIP | Yield |
| 1 | 1 equiv. | — | 37% |
| 2 | 1 equiv. | 0.2 equiv. | 66% |
| 3 | 0.5 equiv. | 0.2 equiv. | 41% |

The tandem oxidation/oxidative esterification reactions of 1a both with and without HFIP were also monitored for 10 h by GC analysis (Fig. 1). In the presence of HFIP, alcohol 1a was converted to ester 1b (76% conversion at 10 h); neither aldehyde 1c nor HFIP-ester9 was observed during the analysis (Fig. 1a). The amount of TEMPO reduced rapidly at the initial stage (2 h) and then remained almost constant for the remainder of analysis. In the absence of HFIP, the amount of 1b was not greatly increased after 2 h (15% conversion at 10 h). Instead, the amount of cinnamaldehyde was increased. Based on the GC monitoring results, HFIP is assumed to promote the rapid conversion of aldehyde 1c to ester 1b, which is consistent with the results of Table 3. Initially, we speculated that the addition of HFIP helped TEMPO recycling, but the concentration of TEMPO was similar in both reactions with and without HFIP.
 | ||
| Fig. 1 (a) One-pot oxidation/oxidative esterification reaction of 1a to 1b in the presence of hexafluoropropanol (HFIP). (b) One-pot oxidation/oxidative esterification reaction of 1a to 1b in the absence of HFIP. Reaction conditions: 1a (0.5 mmol), 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) (0.5 mmol), IMes (5 mol%), and HFIP (0.1 mmol (a) and 0 mmol (b)) in toluene (0.5 M) at 100 °C. | ||
Next, the tandem oxidation/esterification using independently prepared oxoammonium tetrafluoroborate (BF4−) was investigated (Scheme 1). With 1 equiv. of oxoammonium, 1a underwent oxidation to form aldehyde 1c without forming 1b. Even with 2 equiv. of oxoammonium, 1a was converted to 1c without forming 1b, which indicated that oxoammonium was not an oxidant for this esterification. Furthermore, the reaction of 1c and benzyl alcohol in the presence of oxoammonium did not afford the benzyl ester. These results show that NHC-catalyzed oxidative esterification is promoted by TEMPO radicals rather than by oxoammonium.
 | ||
| Scheme 1 Oxidation of 1a and oxidative esterification of 1c in the presence of oxoammonium. | ||
A reaction mechanism supported by the above-mentioned control experiments and GC studies is proposed in Scheme 2. After cinnamaldehyde 1c was formed by the TEMPO-mediated oxidation of 1a, 1c immediately participated in the carbene-catalyzed oxidative esterification. HFIP is presumed to accelerate the rapid adduct formation of 1c with carbene catalysts, which gives the high yield of esters by lowering the concentration of aldehydes. Intermediates I and II are oxidized by TEMPO radicals via SET mechanism to afford III. As stated in our previous publication,4b,4c intermediate III did not react with TEMPOH to form TEMPO-esters.10 Instead, III reacted with 1a to afford 1b. Reduced TEMPOH was reoxidized by O2.11
 | ||
| Scheme 2 Proposed reaction mechanism. | ||
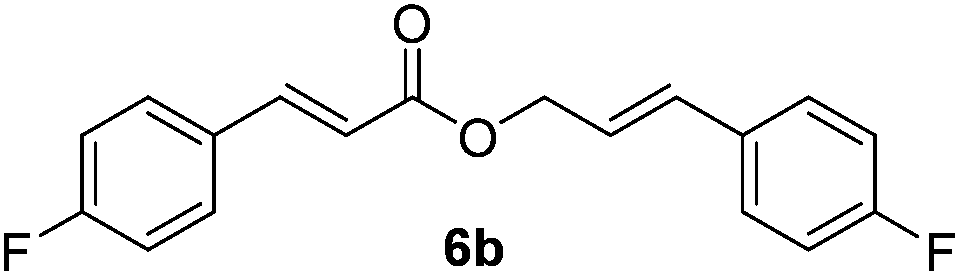
Finally, the substrate scope of this reaction was studied (Table 4). Electron-rich methoxy and methyl-substituted cinnamyl alcohols (2a and 3a) were converted to corresponding esters 2b and 3b in 84% and 73% yield, respectively (entries 1 and 2). Halogen (Cl and Br)-substituted cinnamyl alcohols (4a and 5a) also showed similar reactivity to afford 4b and 5b in 81% and 80% yield, respectively (entries 3 and 4). Electronegative fluoro-substituted cinnamyl alcohol 6a was converted to 6b in a lower yield (58%, entry 5). Electron-deficient NO2-substituted cinnamyl alcohol was also tested under the optimized conditions. Unfortunately, NO2-substituted cinnamyl alcohol was converted to NO2-substituted cinnamaldehyde in 77% yield, and no esterification occurred. In addition to para-substituted compounds, ortho- and meta-substituted compounds 7a and 8a were subjected to the reaction conditions, to afford 7b and 8b in 58% and 68% yield, respectively (entries 6 and 7). Presumably, the steric effect of the substituent affected the yield of this oxidative esterification. Thiophenyl allylic alcohol 9a was transformed to 9b in 63% yield (entry 8). The reaction of aliphatic alcohol 10a afforded the desired product 10b in 26% yield (entry 9). The reaction of 11a showed the steric effect on the olefin of allylic alcohol; the yield of 11b was 23% which is lower than those of cinnamyl alcohols lacking olefin substitution (entry 10).
| Entry | Reactants | Products | Yield |
|---|---|---|---|
| 1 |  |
 |
84% |
| 2 |  |
 |
73% |
| 3 |  |
 |
81% |
| 4 |  |
 |
80% |
| 5 |  |
 |
58% |
| 6 |  |
 |
58% |
| 7 |  |
 |
68% |
| 8 |  |
 |
63% |
| 9 |  |
 |
26% |
| 10 |  |
 |
23% |
Conclusions
We have presented an efficient one-pot oxidation process using NHC catalysts and TEMPO. In this process, TEMPO radicals function as a recyclable oxidant, based on their loadings and GC data. We have also confirmed that our oxidative esterification process proceeded via SET by TEMPO rather than two-electron transfer by oxoammonium. The role of HFIP is to promote the rapid consumption of aldehydes by adduct formation with carbene catalysts, resulting in high yields of oxidative esterification. The increased concentration of aldehydes is detrimental to carbene-catalyzed oxidative esterification with TEMPO.Acknowledgements
This study was supported by the Korea Research Foundation (no. 2009-0094046 and 2013008819) and the Korea CCS R&D Center (KCRC) grant funded by the Korea Government (Ministry of Education, Science and Technology) (no. 2014M1A8A1049294).Notes and references
- For review articles using nitroxide radicals and oxoammonium salts for oxidation, see: (a) J. M. Bobbitt and M. C. L. Flores, Heterocycles, 1988, 27, 509–533 CrossRef CAS PubMed; (b) A. E. J. de Nooy, A. C. Besemer and H. van Bekkum, Synthesis, 1996, 1153–1174 CrossRef CAS PubMed; (c) R. A. Sheldon, I. W. C. E. Arends, G.-J. T. Brink and A. Dijksman, Acc. Chem. Res., 2002, 35, 774–781 CrossRef CAS PubMed; (d) N. Merbouh, J. M. Bobbitt and C. Brückner, Org. Prep. Proced. Int., 2004, 36, 1–31 CrossRef CAS; (e) R. A. Sheldon and I. W. C. E. Arends, Adv. Synth. Catal., 2004, 346, 1051–1071 CrossRef CAS; (f) T. Vogler and A. Studer, Synthesis, 2008, 1979–1993 CAS; (g) L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 5034–5068 CrossRef CAS PubMed.
- For selected articles of nitroxide-catalyzed alcohol oxidation, see: (a) R. Liu, X. Liang, C. Dong and X. Hu, J. Am. Chem. Soc., 2004, 126, 4112–4113 CrossRef CAS PubMed; (b) G. Pozzi, M. Cavazzini, S. Quici, M. Benaglia and G. Dell'Anna, Org. Lett., 2004, 6, 441–443 CrossRef CAS PubMed; (c) C. I. Herrerías, T. Y. Zhang and C.-J. Li, Tetrahedron Lett., 2006, 47, 13–17 CrossRef PubMed; (d) Y. Xie, W. Mo, D. Xu, Z. Shen, N. Sun, B. Hu and X. Hu, J. Org. Chem., 2007, 72, 4288–4291 CrossRef CAS PubMed; (e) X. Wang, R. Liu, Y. Jin and X. Liang, Chem.–Eur. J., 2008, 14, 2679–2685 CrossRef CAS PubMed; (f) C.-X. Miao, L.-N. He, J.-Q. Wang and J.-L. Wang, Adv. Synth. Catal., 2009, 351, 2209–2216 CrossRef CAS; (g) C.-X. Miao, L.-N. He, J.-L. Wang and F. Wu, J. Org. Chem., 2010, 75, 257–260 CrossRef CAS PubMed; (h) M. Hayashi, M. Shibuya and Y. Iwabuchi, J. Org. Chem., 2012, 77, 3005–3009 CrossRef CAS PubMed.
- For selected articles of alcohol oxidation using oxoammonium salts, see: (a) Z. Ma and J. M. Bobbitt, J. Org. Chem., 1991, 56, 6110–6114 CrossRef CAS; (b) J. M. Bobbitt, J. Org. Chem., 1998, 63, 9367–9374 CrossRef CAS; (c) N. Merbouh, J. M. Bobbitt and C. Brückner, J. Org. Chem., 2004, 69, 5116–5119 CrossRef CAS PubMed; (d) W. F. Bailey and J. M. Bobbitt, J. Org. Chem., 2007, 72, 4504–4509 CrossRef CAS PubMed; (e) C. B. Kelly, M. A. Mercadante, T. A. Hamlin, M. H. Fletcher and N. E. Leadbeater, J. Org. Chem., 2012, 77, 8131–8141 CrossRef CAS PubMed; (f) J. C. Qiu, P. P. Pradhan, N. B. Blanck, J. M. Bobbitt and W. F. Bailey, Org. Lett., 2012, 14, 350–353 CrossRef CAS PubMed; (g) C. B. Kelly, M. A. Mercadante, R. J. Wiles and N. E. Leadbeater, Org. Lett., 2013, 15, 2222–2225 CrossRef CAS PubMed.
- For NHC-catalyzed oxidation of aldehydes using TEMPO, see: (a) J. Guin, S. De Sarkar, S. Grimme and A. Studer, Angew. Chem., Int. Ed., 2008, 47, 8727–8730 CrossRef CAS PubMed; (b) M. Ji, X. Wang, Y. N. Lim, Y.-W. Kang and H.-Y. Jang, Eur. J. Org. Chem., 2013, 7881–7885 CrossRef CAS; (c) M. Ji, S. Lim and H.-Y. Jang, RSC Adv., 2014, 4, 28225–28228 RSC.
- For selected articles of single-electron-transfer by organic reagents, see: (a) J. A. Murphy, T. A. Jhan, S.-z. Zhou, D. W. Thomson and M. Mahesh, Angew. Chem., Int. Ed., 2005, 44, 1356–1360 CrossRef CAS PubMed; (b) F. Schoenebeck, J. A. Murphy, S.-z. Zhou, Y. Uenoyama, Y. Miclo and T. Tuttle, J. Am. Chem. Soc., 2007, 129, 13368–13369 CrossRef CAS PubMed; (c) G. P. McGlacken and T. A. Khan, Angew. Chem., Int. Ed., 2008, 47, 1819–1823 CrossRef CAS PubMed; (d) G. J. Rowlands, Tetrahedron, 2009, 65, 8603–8655 CrossRef CAS PubMed.
- For selected articles of NHC-catalyzed oxidation, see: (a) B. E. Maki and K. A. Scheidt, Org. Lett., 2008, 10, 4331–4334 CrossRef CAS PubMed; (b) C. Noonan, L. Baragwanath and S. J. Cannon, Tetrahedron Lett., 2008, 49, 4003–4006 CrossRef CAS PubMed; (c) B. E. Maki, A. Chan, E. M. Phillips and K. A. Scheidt, Tetrahedron, 2009, 65, 3102–3109 CrossRef CAS PubMed; (d) S. De Sarkar, S. Grimme and A. Studer, J. Am. Chem. Soc., 2010, 132, 1190–1191 CrossRef CAS PubMed; (e) S. De Sarkar and A. Studer, Org. Lett., 2010, 12, 1992–1995 CrossRef CAS PubMed; (f) C. A. Rose and K. Zeitler, Org. Lett., 2010, 12, 4552–4555 CrossRef CAS PubMed; (g) B. Maji, S. Vedachalan, X. Ge, S. Cai and X.-W. Liu, J. Org. Chem., 2011, 76, 3016–3023 CrossRef CAS PubMed; (h) P.-C. Chiang and J. W. Bode, Org. Lett., 2011, 13, 2422–2425 CrossRef CAS PubMed; (i) E. E. Finney, K. A. Ogawa and A. J. Boydston, J. Am. Chem. Soc., 2012, 134, 12374–12377 CrossRef CAS PubMed; (j) P. Arde, B. T. Ramanjaneyulu, V. Reddy, A. Saxena and R. V. Anand, Org. Biomol. Chem., 2012, 10, 848–851 RSC; (k) T. Uno, T. Inokuma and Y. Takemoto, Chem. Commun., 2012, 48, 1901–1903 RSC; (l) I. N. C. Kiran, K. Lalwani and A. Sudalai, RSC Adv., 2013, 3, 1695–1698 RSC; (m) L. Möhlmann, S. Ludwig and S. Blechert, Beilstein J. Org. Chem., 2013, 9, 602–607 CrossRef PubMed; (n) J. Zhao, C. Mück-Lichtenfeld and A. Studer, Adv. Synth. Catal., 2013, 355, 1098–1106 CrossRef CAS; (o) S. De Sarkar, A. Biswas, R. C. Samanta and A. Studer, Chem.–Eur. J., 2013, 19, 4664–4678 CrossRef CAS PubMed.
- (a) J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2005, 127, 10840–10841 CrossRef CAS PubMed; (b) B. E. Maki, A. Chan, E. M. Phillips and K. A. Scheidt, Org. Lett., 2007, 9, 371–374 CrossRef CAS PubMed; (c) N. A. Owston, A. J. Parker and J. M. J. Williams, Chem. Commun., 2008, 624–625 RSC; (d) T. Zweifel, J.-V. Naubron and H. Grützmacher, Angew. Chem., Int. Ed., 2009, 48, 559–563 CrossRef CAS PubMed; (e) B. Xu, X. Liu, J. Haubrich, R. J. Madix and C. M. Friend, Angew. Chem., Int. Ed., 2009, 48, 4206–4209 CrossRef CAS PubMed; (f) H. Miyamura, T. Yasukawa and S. Kobayashi, Green Chem., 2010, 12, 776–778 RSC; (g) S. Gowrisankar, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 5139–5143 CrossRef CAS PubMed; (h) C. Liu, J. Wang, L. Meng, Y. Deng, Y. Li and A. Lei, Angew. Chem., Int. Ed., 2011, 50, 5144–5148 CrossRef CAS PubMed; (i) P. Karthikeyan, S. A. Aswar, P. N. Muskawar, P. R. Bhagat and S. S. Kumar, Catal. Commun., 2012, 26, 189–193 CrossRef CAS PubMed.
- For NHC-catalyzed ester synthesis from alcohols, see: Y. Oda, K. Hirano, T. Satoh, S. Kuwabata and M. Miura, Tetrahedron Lett., 2011, 52, 5392–5394 CrossRef CAS PubMed.
 .
.- In our previous publication (ref. 4b), the independently prepared TEMPO-ester was exposed to our reaction conditions including carbene catalysts, resulting in the recovery of TEMPO-ester and no desired products formed.
- A. Dijkśman, I. W. C. E. Arends and R. A. Sheldon, Org. Biomol. Chem., 2003, 1, 3232–3237 Search PubMed.

Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental and spectral analysis for new compounds. See DOI: 10.1039/c4ra08133a |
| This journal is © The Royal Society of Chemistry 2014 |