
Ring-opening metathesis polymerization using polyisobutylene supported Grubbs second-generation catalyst†
Mohammed Al-Hashimi*a,
M. D. Abu Bakara,
Khaled Elsaidb,
David E. Bergbreiterc and
Hassan S. Bazzi*a
aDepartment of Chemistry, Texas A&M University at Qatar, P.O. Box 23874, Doha, Qatar. E-mail: mohammed.al-hashimi@qatar.tamu.edu; bazzi@tamu.edu
bDepartment of Chemical Engineering, Texas A&M University at Qatar, P.O. Box 23874, Doha, Qatar
cDepartment of Chemistry, Texas A&M University, P.O. Box 30012, College Station, Texas 77842-3012, USA
First published on 2nd September 2014
Abstract
Olefin metathesis is among the most powerful tools for the formation of regio- and steroselective carbon–carbon double bonds. Applying the principles of Green Chemistry to the syntheses of polymers by developing useful strategies to facilitate catalyst and polymer product separation after a polymerization is vital. In the present study, a phase selectively soluble polymer bound second generation Grubbs catalyst was successfully used to carry out ring-opening metathesis polymerization (ROMP) on norbornene and a variety of different exo-norbornene derivatives. Polymers with low ruthenium contamination levels were achieved in comparison to the non-supported Grubbs catalyst which required multiple precipitations. Furthermore, the bound catalyst exhibits similar catalytic activity to its homogenous counterpart.
Introduction
The development of well-defined and highly active ruthenium metathesis catalysts having excellent functional group tolerance has been fundamental to the widespread application of olefin metathesis,1–4 in particular for the synthesis of natural products and small molecules.5Ring-opening metathesis polymerization (ROMP) has become an extremely valuable tool for the preparation of functional homopolymers6 providing a wide range of polymers with unique architectures.7 Grubbs (G1, G2 and G3) and Hoveyda–Grubbs (HG1 and HG2) ruthenium based catalysts are among the widely used complexes in both academia and industry (Fig. 1).8–10 Despite these advances a relatively high price of these catalysts and the removal of Ru from the reaction product predominantly in the pharmaceutical industry represent an important disadvantage for their practical application.11
 | ||
| Fig. 1 Ruthenium-based olefin metathesis catalysts. | ||
The development of supported Ru catalysts for olefin metathesis has attracted increasing interest, both from the view of environmental (removal of heavy metal impurities) and economic benefit (high ruthenium cost). We12–14 and several research groups15–26 have reported the use of polymeric and solid supports for immobilizing ruthenium based catalysts. Among the several approaches the majority of articles report on using supported Grubbs second generation catalysts G2 for ring closing metathesis (RCM). Barrett et al. were the first to report the immobilization of G2 on vinyl modified poly(styrene) beads, the so called “boomerang” polymer supported catalyst was used up to three times in RCM almost without loss of any activity.27 Blechert et al. reported the synthesis of supported G2 on poly(styrene) Merrifield resin, the catalyst was used in RCM, cross and enyne metathesis affording near quantitative conversions.15 Fürstner et al. prepared hydroxyalkyl-functionalized G2 complex and was shown to catalyze a serious of RCM. The catalyst was recycled up to three times, but it showed lower catalytic activity than its homogenous counterpart.28 Weck et al. utilized poly(norbornene) as a support for Grubbs second generation catalyst. The reactivity of the supported NHC ruthenium complex were studied for RCM, the authors reported good conversions yielding Ru free products.29
Buchmeiser's group are among the few that have reported the use of supported G2 catalyst for ROMP. They used a monolithic support to immobilize G2 complex, the catalyst was easily removed by filtration at the end of the reaction. Reasonable yields and high activity were obtained in both RCM and ROMP, while the ruthenium residue in the products for RCM was found to be 70 ppm.16 Grubbs et al. published the use of polyethylene glycol (PEG) supported G2 complex. The water soluble catalyst efficiently carried out the ROMP of norbornene derivatives, however, low turnover numbers (TONs) for RCM was reported.
In order to expand the scope of our work using soluble supports we now wish to report the synthesis of polyisobutylene supported Grubbs second-generation catalyst and its application in ring-opening metathesis polymerization.
Results and discussions
Synthesis of soluble PIB-supported 2nd generation Ru complex 6 and characterization
Recently, we elaborated a simple synthetic protocol for preparation of polyisobutylene (PIB)-supported N-heterocyclic carbene (NHC) ligand 5.12 Using this methodology we synthesized the new soluble PIB-supported 2nd generation Ru complex 6. The synthetic routes to complex 6 are outlined in Scheme 1. Accordingly, to a solution of N-heterocyclic carbene ligand 5 in THF at room temperature was added potassium tert-butoxide, resulting in the deportation of the corresponding NHC salt to form the free carbene. In situ the free carbene was reacted with Cl2Ru(CHPh)(PCy3)2 G1 in toluene at room temperature and the reaction was heated to 80 °C. After purification a waxy brown-pink oil in 60% yield was obtained. | ||
| Scheme 1 Synthesis of PIB-supported second-generation Grubbs complex 6. | ||
The structure of complex 6 was verified by 1H NMR, 31P NMR and 13C NMR spectroscopy (Fig. 2). As one might expect, the 1H NMR spectrum of complex 6 (Fig. 2a) shows a shift for the benzylidene proton peak from δ = 20.02 ppm (shift for the first-generation Grubbs G1 complex) to δ = 19.05 ppm, consistent with the parent none supported 2nd generation Grubbs complex G2. However, at first 31P NMR spectroscopy indicated that some triphenyl phosphine oxide was still present 31P δ = 50.04 ppm. Using the heptane-phase selective solubility of complex 6 due to the polyisobutylene-containing ligand to our advantage a liquid/liquid separations using heptane/methanol was introduce which ultimately resulted in removal of the phosphine oxide affording the pure complex as depicted in Fig. 2b.
 | ||
| Fig. 2 NMR spectra (1H and 31P NMR) of PIB supported complex 6. | ||
The reaction was first evaluated using kinetic studies at room temperature in deuterated THF (0.5 mL) using 1 mol% catalyst. Conveniently the polymerizations were conducted in NMR tubes at a spinning rate of 20 Hz. The conversion of monomer M7 to polymer P8 was monitored using 1H NMR spectroscopy (Fig. 3a and b). Interestingly, a marked difference in reactivity profiles is observed for complex 6 and G2. The NB M7 was 93% converted to polynorbornene P8 using complex 6 in 60 min resulting in a colorless solution of polynorbornene P8 with low viscosity. In comparison to the ROMP of NB with the parent Grubbs complex G2, complex 6 exhibited a faster ROMP initiation with greater than ca. 70% conversion occurring at 25 °C within the first 10 min (Fig. 3c) and progressed to ca.90% in 30 min. Whereas initiation was significantly slower with the non-supported Grubbs complex G2 ca.31% conversion was achieved after 10 min and ca.76% after 30 min. However, 99% conversion was achieved within a 60 min. Both complexes G2 and 6 achieved the same conversion in the 46 min 92%. Increasing the catalyst loading of complex 6 to 1.5 mol% results in 99% conversion after 20 min. The results for the kinetic studies obtained using complex G2 agree well with previous published data in the literature.30
 | ||
| Fig. 3 Rates of conversion of NB monomer to polynorbornene-complexes G2 and 6. | ||
The trans–cis double bond ratio in the polymer chain was determined from 1H NMR spectroscopy as depicted in Fig. 4. The olefinic peaks of the monomer at δ = 5.85 ppm are replaced by new signals at δ = 5.26 and 5.09 ppm, which correspond to the cis and trans double bonds of the polymer, respectively. In accord with previous reports, both complexes resulted in polymer P8 having a slight excess of trans ratio (trans![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cis ∼ 59:41 complex G2, 51:49 complex 6).31
:cis ∼ 59:41 complex G2, 51:49 complex 6).31
 | ||
| Fig. 4 1H NMR spectra in THF-d8 of trans–cis double bond of polymer P8 using supported and non-supported Ru complex. | ||
A slight difference in the trans–cis double bond ratio was found between the polymers prepared using the PIB supported Ru complex and non-supported complex (Table 1). The ratio of the trans double bond in polymer P8 prepared with the supported complex was lower. This small difference in stereoselectivity corresponds to a ca. 0.17 kcal mol−1 difference in activation energy for the stereoselectivity in trans–cis isomer formation for catalyst G2 versus 6. Accounting for such small differences is necessarily speculative. One possibility is that there is some sort of steric effect of the PIB group. However, other studies by our group have shown that terminally functionalized polymer supports have no significant steric effect.32–35
| Entrya | Polymers | Product yieldc (%) | Mnd (g mol−1) | PDId | Trans:cis |
Ru contente | |
|---|---|---|---|---|---|---|---|
| (%) | ppm | ||||||
| a 1 mol% catalyst.b Using non-supported Grubbs 2nd generation complex G2.c Isolated yield.d Molecular weight and PDI were determined by GPC.e Measurements were done using ICP-MS analysis, the mol% Ru is the % leached into the polymer. | |||||||
| 1 | P8 | 80 | 488900 |
2.79 | 51:49 |
3.45 | 464 |
| 2 | P8b | 81 | 456400 |
2.85 | 59:41 |
46.4 | 6134 |
| 3 | P11 | 78 | 345900 |
1.71 | 52:48 |
2.09 | 130 |
| 4 | P12 | 63 | 821600 |
1.83 | 46:54 |
0.98 | 71 |
| 5 | P13 | 60 | 1700000 |
1.11 | 44:56 |
1.82 | 139 |
| 6 | P13b | 71 | 125500 |
2.34 | 46:54 |
16.6 | 1083 |
| 7 | P14 | 80 | 737600 |
2.0 | 43:57 |
3.41 | 156 |
| 8 | P15 | 74 | 163800 |
2.16 | 44:56 |
6.1 | 252 |
| 9 | P17 | 65 | Insoluble | — | — | 9.25 | 543 |
| 10 | P19 | 60 | Insoluble | — | — | 4.8 | 361 |
Moreover, prior work did not show any significant difference in trans–cis isomer ratio for polymerizations of a functionalized norbornene using a low molecular weight or a PIB-bound Grubbs–Hoveyda ROMP catalyst.12 We speculate that the PIB groups that are associated with the active Ru alkylidene chain growth site may produce a local solvent effect wherein they interact with this relatively hydrophobic polynorbornene chain. Even a modest such effect could easily account for the observed small change in stereoselectivity for the non-supported complex G2 versus the PIB-supported complex 6.
The reaction mixture containing the polymer and Ru catalyst residue in THF was concentrated in vacuo then the Ru residue was separated by dissolving the crude mixture in minimum amount of DCM and precipitating the mixture in hexane. Thus, the homoploymer was isolated as a white solid, while the PIB–Ru complex remains in the hexane layer. The results obtained by GPC analysis show that the number average molecular weight (Mn) of 488900 g mol−1 with a monomodal polydispersity index (PDI) of 2.79, while using the non-supported Ru resulted in a polymer having an Mn of 456400 g mol−1 and PDI 2.85 as depicted in Table 1.
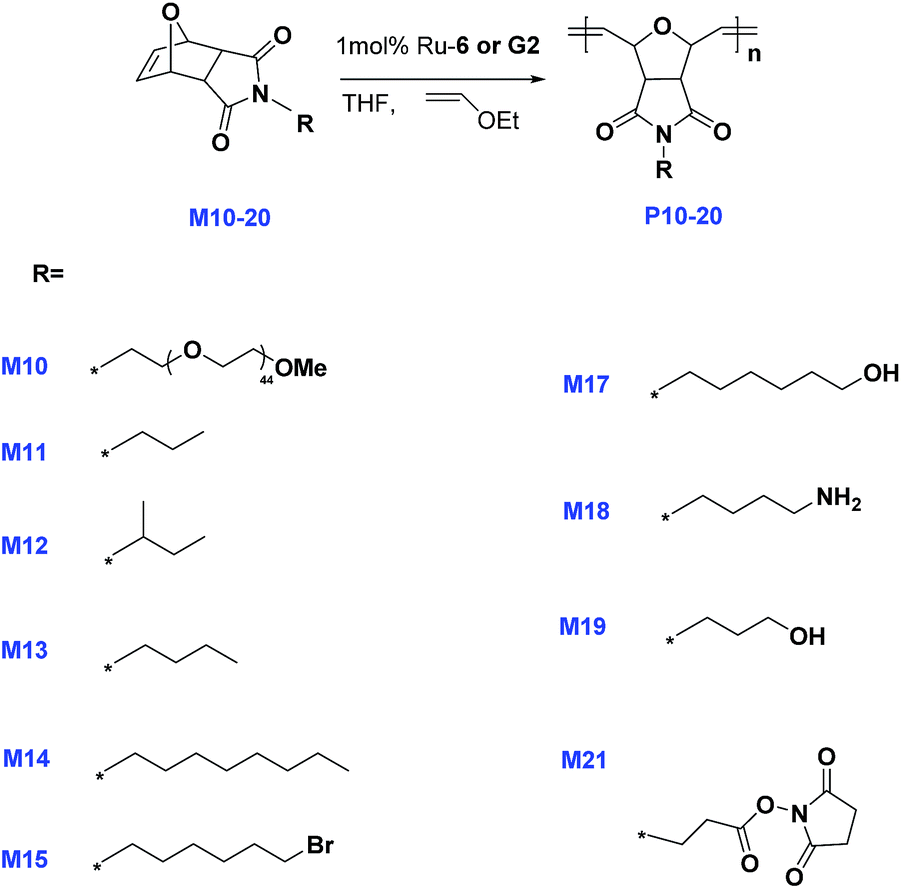
Encouraged by the promising result and to examine the scope of complex 6 we decided to investigate the performance of the new complex on a wider range of substrates. Substituted exo-norbornene imides were selected as the monomers due to their ability to undergo living ROMP, as well as their ability to be easily functionalized. We therefore prepared a variety of monomers from exo-oxabicyclo-[2.2.1]-hept-5-ene-2,3-dicarboximide M9. Monomers M11–M14 were synthesized according to our previously reported method12 Mitsunobu coupling of imide M9 with methoxy-terminated poly(ethylene glycol) (average Mw = 2000) afforded monomer M10 (Scheme 2). Monomers M17–M21 illustrated in Scheme 2 were prepared according to the reported procedure with slight modifications.36 Reaction of exo-anhydride M16 with the appropriate diamine(butane-1,4-diamine) or amino alcohol, 6-amino-1-hexanol and 3-amino-1-propanol afforded monomer M17, M18 and M19 in 50–60% yield. Oxidation of the primary alcohol M19 with CrO3 afforded the carboxylic acid derivative M20, respectively. Coupling of monomer M20 with N-hydroxysuccinimide (NHS) in the presence of 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC) afforded the ester M21.
 | ||
| Scheme 2 Synthetic pathway for the synthesis of ROMP Monomers. | ||
The reactivity of the PIB supported complex 6 with the appropriate ROMP substrates M10 to M20 was evaluated (Scheme 3). The reactions were carried out using 1 mol% complex 6 under rigorously inert conditions in dry THF at room temperature. The rate of monomer conversion was monitored using 1H NMR spectroscopy recorded in CDCl3. Monomers M11 to M15 were fully converted to the desired polymers P11–P15 within 1 h, wherein resonances at δ 6.53 ppm for the cyclic olefin were not observed. Interestingly, complex 6 exhibits very low activity for the ROMP of M10, M18 and M20, showing just 7% conversion after 1.5 h at 25 °C in THF. Even after the reaction time was extended to 24 h this showed no particular change in the conversion. We infer the low monomer conversion due to the decomposition of the Ru complex, yielding the phosphonium oxide as the sole 31P product as analyzed by NMR spectroscopy. Furthermore, interestingly, M17 and M19 resulted in formation of a white solid precipitate within minutes. This was found to be essentially insoluble in common organic solvents, Moreover, the analysis of the polymers by NMR spectroscopy and GPC was made very difficult.
 | ||
| Scheme 3 Synthesis of homo-polymers via ROMP. | ||
Homopolymers P11–15 were separated from the Ru residue as described previously. GPC analysis of polymers P11–15 showed broad PDI's 1.2 to 1.9, which is relatively broader in comparison to polymers synthesized by living polymerization using other Grubbs initiators. Previously it has been shown that G2 complex affords polymers with broad polydispersities and high molecular weights, this can be attributed to the basicity of the tricyclohexyl phosphine ligand.30,37 and high catalyst loading.
Inductively coupled plasma mass spectrometry (ICP-MS) analysis was used to determine the content of Ru leaching in the polymer products (Table 1) by dissolving the polymers in concentrated HNO3. As expected the Ru leaching levels are substantially lower when using the supported complex 6 ranging from 0.98% to 3.45%, in comparison to the non-supported complex G2 which had a higher Ru contamination of 16.6% to 46.6% (entries 1 to 8) even after multiple precipitations with hexane. The results compare favorably well with other systems for the removal of ruthenium residues.
Conclusions
In summary, we have synthesized a soluble polymer bound second generation Grubbs Ru complex for ROMP. The supported catalyst can be easily obtained from commercially available starting materials. The catalytic activity of the supported complex is comparable to those of its homogenous counterpart. In addition the supported catalyst combines solution phase chemistry with the ease of separating the Ru form the final product. Importantly, the ruthenium contamination in the product was very low without the need for further purification in comparison to the parent Grubbs catalyst that required multiple precipitations. Currently further investigations in various types of metathesis reactions using the supported Grubbs catalyst is underway.Acknowledgements
The authors gratefully acknowledge support of this work from the Qatar National Research Fund project number: NPRP 4-081-1-016.Notes and references
- A. Furstner, Angew. Chem., Int. Ed., 2000, 39, 3012–3043 CrossRef.
- T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS PubMed.
- Y. Vidavsky, A. Anaby and N. G. Lemcoff, Dalton Trans., 2012, 41, 32–43 RSC.
- S. J. Connon and S. Blechert, Angew. Chem., Int. Ed., 2003, 42, 1900–1923 CrossRef CAS PubMed.
- R. H. Grubbs, Angew. Chem., Int. Ed., 2006, 45, 3760–3765 CrossRef CAS PubMed.
- P. Schwab, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1996, 118, 100–110 CrossRef CAS.
- S. Sutthasupa, M. Shiotsuki and F. Sanda, Polym. J., 2010, 42, 905–915 CrossRef CAS.
- S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168–8179 CrossRef CAS.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–956 CrossRef CAS.
- C. A. Busacca, D. R. Fandrick, J. J. Song and C. H. Senanayake, Adv. Synth. Catal., 2011, 353, 1825–1864 CrossRef CAS PubMed.
- C. Samojlowicz, M. Bieniek and K. Grela, Chem. Rev., 2009, 109, 3708–3742 CrossRef CAS PubMed.
- M. Al-Hashimi, C. Hongfa, B. George, H. S. Bazzi and D. E. Bergbreiter, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 5211–5212 CrossRef CAS PubMed.
- C. Hongfa, H. L. Su, H. S. Bazzi and D. E. Bergbreiter, Org. Lett., 2009, 11, 665–667 CrossRef CAS PubMed.
- C. Hongfa, J. H. Tian, H. S. Bazzi and D. E. Bergbreiter, Org. Lett., 2007, 9, 3259–3261 CrossRef CAS PubMed.
- S. C. Schurer, S. Gessler, N. Buschmann and S. Blechert, Angew. Chem., Int. Ed., 2000, 39, 3898 CrossRef CAS.
- M. Mayr, B. Mayr and M. R. Buchmeiser, Angew. Chem., Int. Ed., 2001, 40, 3839 CrossRef CAS.
- J. Dowden and J. Savovic, Chem. Commun., 2001, 37–38 RSC.
- S. J. Connon, A. M. Dunne and S. Blechert, Angew. Chem., Int. Ed., 2002, 41, 3835–3838 CrossRef CAS.
- S. Varray, R. Lazaro, J. Martinez and F. Lamaty, Organometallics, 2003, 22, 2426–2435 CrossRef CAS.
- N. Audic, H. Clavier, M. Mauduit and J. C. Guillemin, J. Am. Chem. Soc., 2003, 125, 9248–9249 CrossRef CAS PubMed.
- Q. W. Yao and A. R. Motta, Tetrahedron Lett., 2004, 45, 2447–2451 CrossRef CAS PubMed.
- S. T. Nguyen and R. H. Grubbs, J. Organomet. Chem., 1995, 497, 195–200 CrossRef CAS.
- M. Ahmed, A. G. M. Barrett, D. C. Braddock, S. M. Cramp and P. A. Procopiou, Tetrahedron Lett., 1999, 40, 8657–8662 CrossRef CAS.
- S. Zaman and A. D. Abell, Tetrahedron Lett., 2009, 50, 5340–5343 CrossRef CAS PubMed.
- S. Cetinkaya, E. Khosravi and R. Thompson, J. Mol. Catal. A: Chem., 2006, 254, 138–144 CrossRef CAS PubMed.
- B. H. Lipshutz and S. Ghorai, Tetrahedron, 2010, 66, 1057–1063 CrossRef CAS PubMed.
- M. Ahmed, T. Arnauld, A. G. M. Barrett, D. C. Braddock and P. A. Procopiou, Synlett, 2000, 1007–1009 CAS.
- S. Pruhs, C. W. Lehmann and A. Furstner, Organometallics, 2004, 23, 280–287 CrossRef.
- W. J. Sommer and M. Weck, Adv. Synth. Catal., 2006, 348, 2101–2113 CrossRef CAS PubMed.
- R. Tuba, R. C. da Costa, H. S. Bazzi and J. A. Gadysz, ACS Catal., 2012, 2, 155–162 CrossRef CAS.
- C. W. Bielawski, D. Benitez, T. Morita and R. H. Grubbs, Macromolecules, 2001, 34, 8610–8618 CrossRef CAS.
- D. E. Bergbreiter and R. Chandran, J. Org. Chem., 1986, 51, 4754–4760 CrossRef CAS.
- D. E. Bergbreiter and R. Chandran, J. Am. Chem. Soc., 1987, 109, 174–179 CrossRef CAS.
- D. E. Bergbreiter and Y. C. Yang, J. Org. Chem., 2010, 75, 873–878 CrossRef CAS PubMed.
- C. Hongfa, J. H. Tian, J. Andreatta, D. J. Darensbourg and D. E. Bergbreiter, Chem. Commun., 2008, 975–977 RSC.
- N. B. Sankaran, A. Z. Rys, R. Nassif, M. K. Nayak, K. Metera, B. Z. Chen, H. S. Bazzi and H. F. Sleiman, Macromolecules, 2010, 43, 5530–5537 CrossRef CAS.
- J. Vargas, E. S. Colin and M. A. Tlenkopatchev, Eur. Polym. J., 2004, 40, 1325–1335 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, ICP-MS Digestion Procedure, NMR characterization, and assignment of polymers. See DOI: 10.1039/c4ra08046g |
| This journal is © The Royal Society of Chemistry 2014 |