
Recent progress in highly efficient Ag-based visible-light photocatalysts
Abstract
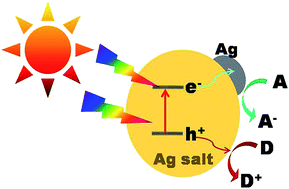
As one of the most promising and efficient approaches for remediating the deterioration of natural environments, semiconductor-based photocatalysis has received considerable attention. To date, numerous efforts have been focused to explore novel materials for highly efficient photocatalysis under visible light or sunlight irradiation. Among them, Ag-based compounds are emerging to be a promising candidate because of their excellent visible light-responsive photoelectrochemical properties. This review summarizes the recent progress in the design and fabrication of Ag-compound-based semiconductor photocatalysts and their applications in the photocatalytic decomposition of organic molecules. Initially, the mechanisms of the related photocatalytic reactions will be discussed, and then we will highlight some of the recent progresses in Ag-based micro- or nano-structured material fabrication that exhibit enhanced photocatalytic performance. These novel and highly efficient photocatalysts mainly include Ag2O, Ag2S, AgX (X = Cl, Br, I), Ag2CO3 and Ag3PO4. We expect that the present tutorial review will provide insights in the direction of the future visible-light photocatalyst design.
 Please wait while we load your content...
Please wait while we load your content...

