
Organic amine-functionalized silica-based mesoporous materials: an update of syntheses and catalytic applications
Dharitri Rath
a,
Surjyakanta Rana
b and
K. M. Parida
*a
aCenter for Nano Science and Nano Technology, Department of Chemistry, Institute of Technical Education & Research, Siksha ‘O’ Anusandhan University, Khandagiri, Bhubaneswar-751030, OR, India. E-mail: kulamaniparida@soauniversity.ac.in
bSchool of Chemistry & Physics, College of Agriculture, Engineering & Science, University of KwaZulu-Natal, Westville Campus, Durban 4000, South Africa
First published on 14th October 2014
Abstract
Nowadays, inorganic–organic hybrid materials having pores in the mesoporous range are an intensively studied new category of demanding materials. By template synthesis, the coupling of inorganic and organic components gives pore sizes between 2 and 15 nm with very high surface area. The inorganic–organic hybrid materials were prepared in two ways: one was by co-condensation and the other by post-synthesis method. The inorganic part provides mechanical strength and the organic part shows functional activities. This review gives an overview of the preparation, properties, and potential applications of these materials in the areas of adsorption of pollutant gases like CO2 and heavy metals and in catalysis. Their activity is found to be very impressive in all these fields and it is hoped to be improved in the near future.
 Dharitri Rath | Dr Dharitri Rath joined the Institute of Minerals and Materials Technology in 2005 as a junior research fellow. She completed her PhD in 2011 under the guidance of Dr K. M. Parida from Utkal University, Odisha. Now she is working as Asst Professor in the Department of Chemistry, Institute of Technical Education and Research, Siksha 'O' Anusandhan University. She is the author of 11 international journal papers and one book chapter. Her research interests focus on metal-modified mesoporous materials, functionalized mesoporous materials and their applications in catalytic and photocatalytic reactions. |
 Surjyakanta Rana | Dr Surjyakanta Rana was born in 1982 and completed his PhD degree at the Department of Chemistry, Utkal University, Odisha, India in 2013 under the supervision of Dr K. M. Parida. He is the author of 15 international journal papers and one book chapter. His research interest focuses on functionalized mesoporous silica and its catalytic applications. |
 K. M. Parida | Dr K. M. Parida is working as Professor in Chemistry and Director in the Centre for Nano Science and Nano Technology, Institute of Technical Education & Research, Siksha ‘O’ Anusandhan University, Bhubaneswar, Odisha. He is the author of more than 270 international journal papers and 18 national and international patents. His research interest focuses on the design and development of materials comprising a wide cross section such as metal oxides, metal phosphates, metal sulfates, cationic and anionic clays, perovskites, zeolites, nano metal/metal oxide/complex promoted mesoporous materials, naturally occurring materials such as manganese nodules, manganese nodule leached residues, and manganese oxides of natural origin for application in catalysis and photocatalysis in the sector of energy and environment. |
1. Introduction
Many porous materials have been widely studied with regard to technical applications as excellent adsorbents and heterogeneous catalysts. According to the IUPAC classification, porous materials are divided into three classes: microporous (pore size < 2 nm), mesoporous (2–50 nm),1,2 and macroporous (>50 nm) materials3 (Fig. 1). | ||
| Fig. 1 Types of porous materials depending on their pore sizes. | ||
The mesoporosity of silica-based materials has come to the forefront as a new and exciting research field of great scientific and technological importance in heterogeneous catalysis. The ability to design both the size and wall surface characters of the pores is important towards imposing a framework for tailoring and fine-tuning catalytic activities and optoelectronic properties of further embedded clusters.4,5 It has become an exciting field to be explored simultaneously by chemists, physicists, and engineers.
2. Mesoporous materials
The term zeolite was first used by the Swedish mineralogist Axel Fredrik Cronstedt in 1756. Zeolites are crystalline and microporous aluminosilicate minerals. A defining feature of zeolites is that their frameworks are made up of four-connected networks of atoms. Just like a tetrahedron with a silicon atom in the middle and four oxygen atoms at the corners. These tetrahedra can then link together by their corners to form a rich variety of beautiful structures. The general formula of zeolite is Mx/n[(AlO2)x(SiO2)y]ZH2O, where M represents a charge-compensating cation with valency n. The ratio y/x may have any value ranging from one to infinity. Z is the number of water molecules, which can be reversibly adsorbed and desorbed into the zeolite micropores.Even though zeolites, having pore dimensions of 5 to 7 Å, serve the purpose of many industrial reactions by providing high surface area, the pore dimensions are not sufficient to accommodate a broad spectrum of large molecules. The performance of zeolite systems is limited by diffusion constraints associated with smaller pores. So it is the aim of industrial and scientific research to expand the pore size of zeo-type materials from the microporous to the mesoporous range.
In 1990, Yanagisawa et al.6 reported the synthesis of mesoporous materials characteristic of MCM-41 through intercalation of long-chain (typically C16) alkyltrimethylammonium cations into the layered silicate kanemite, followed by calcination to remove the organic species. Unfortunately no further characterization data were available and therefore Yanagisawa's results have been ignored.
Typical mesoporous materials include some kinds of silica and alumina that have fine mesopores of similar size. Mesoporous oxides of niobium, titanium, tantalum, zirconium, cerium and tin have also been reported.
2.1 Silica-based mesoporous materials
The pore size constraint (15 Å) of microporous zeolite was overcome with the first successful report on the synthesis of mesoporous materials (M41S) by Mobil researchers, with well-defined pore sizes of 20–500 Å. The high surface area and precise tuning of the pores are among the desirable properties of these materials. These materials are mainly used in a new synthetic approach where, instead of a single molecule as templating agent as in the case of zeolites, self-assemblies of molecular aggregates or supramolecular assemblies are employed as templating agent. They have a uniform hexagonal arrangement of pores with size ranging between 1.5 and 10 nm.7 This material was named Mobil Composition of Materials no. 41 or MCM-41.8 Researchers at the University of California in Santa Barbara produced silica nanoparticles with much larger pores of size 4.6 to 30 nm.9 This material was named Santa Barbara Amorphous type material or SBA-15. In this material particles are also arranged in a hexagonal array. Yanagisawa and co-workers6 prepared another mesoporous silica from kanemite (NaHSi2O5·3H2O) and CTA cation designated as FSM-16. FSM-16 is structurally similar to but functionally different from MCM-41. It shows better adsorption properties and surface chemistry. MCM-48 is another mesoporous material with a cubic structure and a three-dimensional pore system. The two other phases of mesoporous silica are lamellar (MCM-50) and molecular organic octomer (a surfactant–silica composite) and are unstable (Fig. 2).10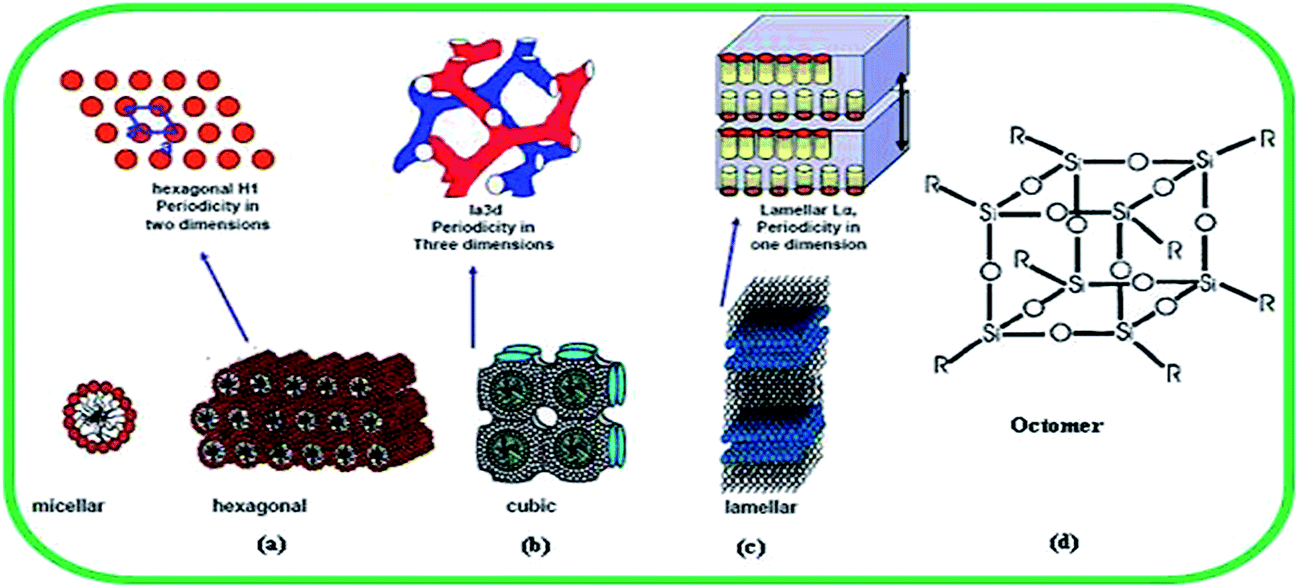 | ||
| Fig. 2 Structures of mesoporous silica materials: (a) MCM-41 (hexagonal), (b) MCM-48 (cubic), (c) MCM-50 (lamellar), and (d) octomer. Reproduced from ref. 10. “Reprinted with permission from (P. Selvam, S. K. Bhatia, C. G. Sonwane, Ind. Eng. Chem. Res., 2001, 40, 3237). Copyright (2014) American Chemical Society”. | ||
In 2007, a new porous material was formulated by Nandiyanto et al. in Hiroshima University. They prepared the mesoporous material by a simple sol–gel method11 with controllable pore size from 3 to 15 nm and outer diameter from 20 to 100 nm. This material was named Hiroshima Mesoporous Material, and abbreviated as HMM. These materials show excellent adsorption properties for large molecules.12 Physical properties of various mesoporous silica materials are shown in Table 1.
| Sample code | Structural data | Mean pore size (nm) | Reference | |
|---|---|---|---|---|
| Dimensionality, crystal system, space group | Unit cell dimension (nm) | |||
| MCM-41 | 2D hexagonal (P6mm) | a = 4.04 | 3.70 | 7 |
| MCM-48 | Cubic (Ia3hd) | a = 8.08 | 3.48 | 7 |
| FSM-16 | 2D hexagonal (P6mm) | a = 4.38 | 2.80 | 13 |
| SBA-1 | Cubic (Pm3hn) | a = 7.92 | 2.00 | 14 |
| SBA-2 | 3D hexagonal (P63/mmc) | a = 5.40, c = 8.70 | 2.22 | 15 |
| SBA-3 | 2D, hexagonal (P6mm) | a = 4.75 | 2.77 | 14 |
| SBA-8 | 2D rectangular (cmm) | a = 7.57, b = 4.92 | 1.87 | 16 |
| SBA-11 | Cubic (Pm3hm) | a = 10.64 | 2.50 | 17 |
| SBA-12 | 3D hexagonal (P63/mmc) | a = 5.40, c = 8.70 | 3.10 | 17 |
| SBA-14 | Cubic (Pm3hn) | a = 4.47 | 2.40 | 17 |
| SBA-15 | 2D hexagonal (p6mm) | a = 11.6 | 7.80 | 18 |
| SBA-16 | Cubic (Im3hm) | a = 17.6 | 5.40 | 17 |
| HMM | 2D hexagonal (P6mm) | a = 5.70 | 3.10 | 19 |
2.2 Mechanistic pathways for the formation of silica-based mesoporous materials
A variety of synthetic procedures can be adopted for the preparation of mesoporous silica. However, there is one thing all these procedures have in common next to the obvious presence of a source of silica: a templating agent. A template is a structure-directing agent, which is usually a relatively simple molecule or ion, around which a framework is built up. Due to their large hydrophobic alkyl chains the template ions will aggregate together in order to minimize energetically unfavorable interactions of the apolar alkyl chains with the very polar water solvent molecules. The resulting aggregates of ions are called micelles. It follows that these micelles have a hydrophobic core, containing the large alkyl chains, and a hydrophilic surface, due to the ionic character of the ammonium head groups.Spherical geometry is the mostly favorable form of micelles because in this geometry the surface energy is minimized most efficiently. Moreover, this confirmation allows the largest number of micelles to be formed out of a certain amount of template, which is attractive considering the entropy of the system.
The silica source and nature of surfactant decide the nature of interaction as illustrated in Fig. 3. By varying the synthesis conditions like the silica source and the type of surfactant used, many other mesoporous materials can be synthesized.20–22 In addition to the co-operative pathway, which is discussed above, also the true liquid crystal templating pathway23 and nano-casting are used to form ordered mesoporous materials as hard templates.24,25
 | ||
| Fig. 3 Basic interaction between silica and surfactant. Reproduced from ref. 10. “Reprinted with permission from (P. Selvam, S. K. Bhatia, C. G. Sonwane, Ind. Eng. Chem. Res., 2001, 40, 3237). Copyright (2014) American Chemical Society”. | ||
In this method there must be an attractive interaction between the silica source and the template, in which the structure-directing agent (SDA) is added without any change in its phase. The various interactions between the inorganic precursors and the head groups of the SDA are shown in Fig. 3. Huo et al.21,22 suggested four possible interactions between the silica source and the head groups of the SDA. (a) When the SDA has a +ve head group, in basic medium the interaction is represented as S+I− (Fig. 4a; S: surfactant; I: inorganic species). (b) When the SDA has a +ve head group, in acidic medium (below the iso-electric point of Si–OH group) a mediator ion X− (usually a halide) is required to bring about the interaction and is represented as S+X−I+ (Fig. 4b). (c) When the SDA has a −ve head group, in basic medium a mediator metal ion M+ is required to bring about the interaction and is represented as S−M+I− (Fig. 4c). (d) When the SDA has a −ve head group, in acid medium the interaction is represented as S−I+ (Fig. 4d).
 | ||
| Fig. 4 Electrostatic interactions between the inorganic species and the head group of the surfactant in acidic or basic media. Reproduced from ref. 10. “Reprinted with permission from (P. Selvam, S. K. Bhatia, C. G. Sonwane, Ind. Eng. Chem. Res., 2001, 40, 3237). Copyright (2014) American Chemical Society”. | ||
Hence the ruling interactions in pathways (a–d) are of an electrostatic nature. Experimentally it was observed that the surfactant to silica ratio has a significant impact on the composite structure obtained.26 The use of anionic surfactant via S−I+ or S−M+I− interaction results mainly in lamellar and disordered mesostructures which is also demonstrated by Che et al.27,28 The pore size of these materials can be controlled by the length of the alkyl chain of the surfactant used.29 Also the mesopore size can be expanded by the use of some auxiliary organic molecules like organic alkanes30 or fatty acids.31 The negatively charged head group of the anionic surfactant (i.e. palmitic acid or N-lauroyl-L-glutamic acid) interacts with the positively charged ammonium groups of additives for co-condensation of TEOS (tetraethyl orthosilicate). Two alkylammonium surfactants with different alkyl chain length can be mixed to fine-tune the pore size between those of the long- and the short-chain surfactants.32
The proposed mechanistic pathways for the formation of mesoporous silica structures are illustrated in Fig. 5 and 6.7,24,25,33 In the first, the presence of liquid-crystal mesophase prior to the addition of the reagents, i.e., pre-existence of surfactant aggregates (rod-like micelles), followed by the migration and polymerization of silicate anions, results in the formation of the MCM-41 structure.
 | ||
| Fig. 5 (a) Mechanism for the formation of mesoporous materials by structure-directing agents: true liquid-crystal template mechanism, (b) cooperative liquid-crystal template mechanism. Reproduced from ref. 10. “Reprinted with permission from (P. Selvam, S. K. Bhatia, C. G. Sonwane, Ind. Eng. Chem. Res., 2001, 40, 3237). Copyright (2014) American Chemical Society”. | ||
 | ||
| Fig. 6 Proposed mechanistic pathways for layered silica by (1) stacking of silicate surfactant rods, via the formation of an initial (2) lamellar intermediate and (3) silicate bilayer. Reproduced from ref. 10. “Reprinted with permission from (P. Selvam, S. K. Bhatia, C. G. Sonwane, Ind. Eng. Chem. Res., 2001, 40, 3237). Copyright (2014) American Chemical Society”. | ||
The geometry of the micelle formed depends on the concentration of the template used. Initially the geometry is spherical which gradually transforms into long tubes. Finally the tube-like micelles aggregate to form a hexagonal liquid crystalline structure which is the framework for MCM-41. Again on further increasing the template concentration, the hexagonal geometry is transformed to cubic and lamellar structures which form the MCM-48 and MCM-50 materials respectively.21,22,26,27 The liquid crystalline structures act as the actual templates for the different mesoporous materials. The mechanistic pathways are shown in Fig. 6.
3. Scope of the review
Mesoporous silica MCM-41 and various organic amine-modified MCM-41 provide a good base for the dispersal of different organic molecules and nano metal oxides. Moreover the review will discuss detail about the synthesis procedures and widespread applications of these materials. The most important fields are the adsorption of heavy metals and harmful gases and industrially viable organic reactions. This will provide a new outlook to researchers for carrying out work in these fields.4. MCM-41
Among the above mentioned mesoporous silica materials, MCM-41 has an eminent place due to some of its special properties. It is an ordered mesoporous material, displaying a honeycomb-like structure of uniform mesopores running through a matrix of amorphous silica. Because of its structural simplicity and ease of preparation with negligible pore-networking and pore-blocking, it is the most studied among the numerous mesoporous silica materials studied so far. The transmission electron micrograph of MCM-41 depicted in Fig. 7 shows the beautiful honeycomb-like structure. In this figure we can see directly inside the uniform mesopores, which are separated from each other by thin walls of amorphous silica, approximately 1–1.5 nm thick. | ||
| Fig. 7 Transmission electron micrograph of MCM-41. Reproduced from ref. 10. “Reprinted with permission from (P. Selvam, S. K. Bhatia, C. G. Sonwane, Ind. Eng. Chem. Res., 2001, 40, 3237). Copyright (2014) American Chemical Society”. | ||
The most remarkable features of MCM-41, and in general most periodic mesoporous materials, are as follows: well-defined pore shapes (hexagonal/cylindrical), narrow distribution of pore sizes, negligible pore networking or pore blocking effects, very high degree of pore ordering, possibility of tailoring and fine-tuning of the pore dimensions (1.5–20 nm), large pore volumes (>0.6 cm3 g−1), exceptional sorption capacity as a result of the large pore volume, very high surface area (∼700–1500 m2 g−1), large amount of internal hydroxyl (silanol) groups (∼40–60%), high surface reactivity, ease of modification of the surface properties, enhanced catalytic selectivity in certain reactions, and excellent thermal, hydrothermal, chemical, and mechanical stability.
MCM-41 has a very large void fraction, due to the presence of the mesopores, and concomitantly a rather low density. As a result MCM-41 displays a very large specific surface area of approximately 1300 m2 g−1. Therefore, MCM-41 is in principle ideally suited to be used as a support material for heterogeneous catalysts. Moreover, MCM-41 exclusively contains mesopores which can provide access for large molecules and improve diffusion problems, which are frequently encountered in microporous materials such as zeolites.
Unfortunately there is one pronounced drawback associated with MCM-type materials, i.e. limited stability, which is a result of very thin, amorphous pore walls. Because of the very large mesopore surface area, the pore walls are extremely reactive towards a number of agents, resulting in the collapse of the thin walls upon exposure to these agents. The silica-supported materials are not suitable for a large range of processes due to their instability towards steam. Steam either co-fed as diluent or produced during catalysis results in the chemical evaporation of silica. Also, MCM-41 material is unstable towards hydroxide and fluoride solutions as they can dissolve silica. Hence the stability of MCM-41 material is restricted to pH ≤ 7 in aqueous solutions.
4.1 Synthesis of MCM-41
The synthesis of MCM-41 requires four essential components: structure-directing agent (template), solvent, silica source and mineralizing agent. The most commonly used template for MCM-41 is hexadecyl (or cetyl) trimethylammonium bromide (or chloride). It is a template with an alkyl chain containing sixteen –CH2– moieties. This template yields MCM-41 with a uniform pore size of approximately 2.7 nm. By using templates with longer or shorter alkyl chains the pore size can be controlled. Nevertheless, due to the limited range of alkylammonium ions suitable for the preparation of MCM-41, the pore size can be adjusted to a small extent only.34–36 Some auxiliary organics like 1,3,5-trimethylbenzene34,35 can be introduced to adjust the pore size of the material to a remarkable extent. Being apolar, the organics cannot be dissolved in water but they can be absorbed in the hydrophobic core of the template micelles. Due to this absorption the micelles swell, thus increasing the average size of the mesopores in MCM-41 up to values of approximately 8–10 nm in diameter. Mixtures of two surfactants can also be used to fine-tune the pore size of MCM-41. Cheng et al.37 proposed a synthesis procedure for MCM-41 using the C18TABr–C14TABr mixed system. Next to a structure-directing agent and water as a solvent, an important ingredient is a source of silica. Various sources of silica can be used for syntheses, i.e. water glass, amorphous silica and kanemite (a layered silicate structure consisting of anionic silica sheets with charge-compensating sodium ions present in the interlayers). Furthermore, organic silicon alkoxides are also frequently used. The most widely used one is tetraethyl orthosilicate (TEOS).38–52Silica sources can be dissolved in a mineralizing agent like NaOH or concentrated NH4OH or HF. After dissolution of silica, small silicon oxy anions are produced. There is electrostatic attraction between the micelles and the silicate anions and they move towards the micelles. As a result the anion concentration increases on the micelle surface. Again there is repulsive interaction among the silicate ions. In order to assuage the interaction the silicate ions start to condense with each other and form a monolayer of silica over the micelles. At this stage the silica “coated” micelles can start to cluster together by condensation reactions between the silica layers of individual micelles, thus generating the MCM-41 framework. As a result of these processes the pore walls of MCM-41 are amorphous and only 2–3 monolayers thick.53–75
After formation of MCM-41 the pores are filled with the template molecules. These micelles have to be removed to get a perfect mesoporous material. One of the simple methods for template removal is calcination. This is the process of heating a sample in the presence of air. During the removal process the template is decomposed into CO2, NOx and steam. The steam quantity is too low to cause damage to the MCM-41 framework. A schematic representation of this process is shown in Fig. 5.
4.2 Functionalized MCM-41
Siliceous mesoporous materials in neat form lack active sites on their surface. Hence their applications are restricted and their surface has to be modified according to the requirements. In order to utilize the unique properties of the mesoporous materials for specific applications like catalysis, sorption, sensing, ion exchange etc., introduction of reactive organic functional groups is required. The incorporation of organic components as part of the silicate walls or trapped within the channels to form inorganic–organic hybrid materials (IOHMs) remains the main issue. The advantages of IOHMs arise from the fact that the inorganic components can provide mechanical, thermal or structural stability, while the organic features can readily be modified for various specific applications.76–99IOHMs represent the natural interface between the two worlds of chemistry, each with significant contributions and characteristic properties. The advantages and limitations to their field lead to a creative alternative for obtaining new materials with unusual features. The main idea of developing hybrid materials is to take advantage of the best properties of each component and try to decrease their drawbacks, hence resulting in a unique advanced material with potential applications. The possibility of combining the properties of inorganic and organic compounds to get a unique material is a challenging task in recent years. Although we do not know the original birth of hybrid materials exactly, it is clear that mixing of inorganic and organic components was carried out in ancient times. However, it was only at the end of the 20th and the beginning of the 21st century, 7 years after the discovery of Mobil Composition of Materials, that the concept of hybrid materials came to the scene. Then researchers considered IOHMs as innovative and advance materials having potential application in various fields including catalysis.100–112
Functionalized mesoporous oxides possess exceptionally high surface areas, which allow the binding of a large number of surface chemical moieties and hence they can be used as adsorbents and catalysts. Mainly the large diameter and wormhole nature of the pore channels in these oxides are advantageous for the formation of functional materials in which the reactive species are highly dispersed. Hence these are quantitatively accessible for reaction with adsorbate molecules.130–133
A wide range of different materials come under the classification of hybrid materials. These are highly crystalline co-ordination polymers and amorphous sol–gel compounds, with and without interactions between the inorganic and organic units. The most wide ranging definition is that a hybrid material includes two moieties blended on the molecular scale. Commonly, one of these compounds is inorganic and the other one is organic in nature.
The non-calcined silica-based mesophases are regarded as IOHM. However their applications are very limited. These materials are either calcined or solvent-extracted for further applications. Sugi et al.,114 however, found that non-calcined MCM-41 silica exhibits high catalytic activity for the Knoevenagel condensation. From 29Si NMR data, the authors came to the conclusion that the catalytic activity is attributable to basic (SiO)3SiO− sites, which are present in large amounts in non-calcined silica mesophases. According to Tolbert and co-workers,113–115 an ordered silica/surfactant MCM-41 mesophase exhibits remarkable behavior under high pressure. In addition to the as-synthesized amphiphile or silica hybrids, the following materials fit a broad definition of silica-based IOHM: (1) mesoporous silicas with organically modified surfaces, (2) expanded mesoporous silicas, (3) mesoporous organosilicates, and (4) mesoporous silica with occluded organic materials such as polymers.
4.3 Synthetic approaches for functionalized MCM-41
Surface functionalization of inorganic supports with many organic moieties provides organic–inorganic hybrids, where the inorganic and organic components are linked via strong-type interactions (i.e. covalent or iono-covalent bonds). Many research efforts, which have focused on preparing the organic–inorganic hybrids through functionalization of the exterior and/or interior surfaces, prompted the utilization of the resultant materials in the fields of catalysis, separation, sensor design, adsorptions, and nanoscience.According to the location of the organic fragments in the inorganic network, organically modified MCM-41 solids can be divided into three main classes. In the first, the organic groups are rooted to the surfaces of the inorganic walls and protrude into the pores by (i) post-synthetic grafting of X–Si(OR)3 compounds (–X: functional group) to a calcined MCM-41 silica,114,115 (ii) co-condensation of a functionalized organosiloxane with the main source of the inorganic constituent, followed by acid extraction to remove the surfactant molecules from the pores116,117 and (iii) displacement of the surfactant molecules from the mesopores by an organosiloxane through interfacial reactions.118 In the second class, the organic moieties are fused within the silica walls through co-condensation of organosiloxanes of the type (RO)3Si–X–Si(OR)3 (X: organic spacer) with the prime silicon alkoxide, followed by extraction of the surfactant molecules (periodic mesoporous organosilicas, PMO).119–123 Finally, the third class comprises a combination of the aforementioned classes of inorganic–organic hybrids, e.g., some organic groups reside outside and others within the walls of the inorganic network.
 | ||
| Fig. 8 Schematic representation of grafting (post-synthetic functionalization) for organic modification of mesoporous pure silica phases with terminal organosilanes of the type (R′O)3SiR (R = organic functional group). Reproduced from ref. 129. With permission from Copyright © 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, license no. 3472940511823. | ||
The process of grafting is frequently erroneously called immobilization, which is a term that we believe should be reserved for adsorptive methods (e.g., the removal of toxic or environmentally relevant contaminants by adsorbent materials, or the separation of proteins and biocatalysts by restriction of the freedom of movement). Secondary and higher order modifications consist of further reactions of the previously grafted species to create new functionalities.
 | ||
| Fig. 9 Schematic representation of co-condensation method (direct synthesis) for the organic modification of mesoporous pure silica phases. Reproduced from ref. 129. With permission from Copyright © 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, license no. 3472940511823. | ||
 | ||
| Fig. 10 Schematic representation of general synthetic pathway to PMOs that are constructed from bis-silylated organic bridging units. Reproduced from ref. 129. With permission from Copyright © 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, license no. 3472940511823. | ||
Importantly, the nature and the content of organic groups determine the specific properties of these nanocomposites, like the surface hydrophobicity, and hydrothermal, thermal and mechanical properties. However, the uniformity of the organic groups inside the pore channels affects the surface properties, the functionalized organic group reactivity and the accessibility of the porous network for further modifications.
The basicity in Si-MCM-41 molecular sieves is achieved in various ways. One such way is dispersing with alkali metal oxides.130,131 The drawback of this method is that the high pH used in the impregnation may damage the structure. Another way is by modifying the surfaces with organic compounds, particularly amines, by the impregnation method. In this method, the cationic surfactant present in the pores is removed by calcination before functionalization. Recently Kubota et al. used MCM-41 molecular sieve as a catalyst in Knoevenagel condensation reaction at room temperature still containing its structure-directing organic cation: the cetyltrimethylammonium occluded in its pores.132,133 Martins et al. have proposed that the active sites in this catalyst are the basic siloxy SiO− anions and the reaction only occurs at the pore mouths.134,135
The impregnation method for the organic amine aminopropyltriethoxysilane (APTES) typically results in inhomogeneous surface coverage because the introduced organic moieties congregate near the entries to the mesoporous channels and on the exterior surfaces. In the co-condensation method the amine and silica sources are added to the reaction mixture simultaneously.136 The surfactant is removed by acid extraction. In this method the pores of functionalized MCM-41 are occluded by organic amine templates, which provide high activity as a base in mild conditions. The organosilane is more evenly distributed on the silica surface in the case of the co-condensation method than in the case of the post-synthesis method. So the material obtained from the latter method is more efficient as a base catalyst than that obtained from the former. These APTES-modified MCM-41 samples (Fig. 11) show very good catalytic activity towards various base-catalyzed organic transformation reactions such as Knoevenagel condensation, Michael condensation, Henry reaction etc.
 | ||
| Fig. 11 Functionalization of MCM-41 with APTES. | ||
4.4 Amine-functionalized mesoporous silica other than MCM-41
Other mesoporous silica materials are also susceptible to surface modification by organic amines. A lot of work has been done on amine-functionalized SBA-15, SBA-16, MCM-48, silica gel etc. A recent work by Meléndez-Ortiz et al.137 explained the room temperature synthesis of APTES-MCM-48 and its application towards CO2 (ref. 138) and H2S capture in the natural gas sweetening process. MCM-41 was found to be more inclined to pore blocking compared to MCM-48. SBA-15 is among the most attractive mesoporous silicas due to its adjustable pores and thick pore walls. Different amine-modified SBA-15 materials are studied by various research groups. The surface of SBA-15 can be modified with APTES, APTMS, polyethylene imine, polypropylamine and also different primary, secondary and tertiary amines for the adsorption of CO2, NO2, various metal pollutants like Na, K, Ca, and Cu, VOC etc.139–144 Gao et al.145 synthesized a novel kind of spaced amine-modified SBA-15 through Diels–Alder reaction. The condensation of SBA-15-Cp and fumaronitrile was followed by reduction of cyano groups. By this method, the distance between two consecutive amines was found to be same as that of the n-butyl group. The material was found to be an excellent catalyst for Knoevenagel condensation.4.5 Other amine-functionalized oxides
In addition to mesoporous silica, other mesoporous oxides are also able to undergo amine modification. Parida et al. have worked extensively on amine-modified metal oxides like zirconia, titania, titania–silica mixed oxides,146 montmorillonite, layered double hydroxide147 etc. The amine-functionalized zirconia materials were used as effective catalysts for Knoevenagel condensation, Henry reaction and C–S coupling reactions.148–150 The amine-functionalized montmorillonite-supported Cu, Ni catalyst showed synergetic and co-operative effectiveness towards C–S coupling reactions.151,152 Different amine-functionalized TiO2 and ZrO2 can also act as good adsorbents for CO2 and other pollutant gases.153,1545. Applications of amine-modified MCM-41
5.1 Adsorption of CO2
The various amine-functionalized materials act as effective catalysts and adsorbents in several catalytic reactions. Carbon dioxide (CO2) removal is increasingly important because a high CO2 concentration in the atmosphere leads to global climate changes. In addition, the removal of CO2 is also required in cryogenic plants to prevent CO2 solidification. The removal can be achieved by using methods such as liquid absorption, solid adsorption cryogenic techniques, and selective diffusion through polymer, ceramic, or metallic membranes. At present, chemical absorption using liquid amine is commercially used in large-scale separation. However, a relatively high heat of absorption causes a high cost of regenerating primary and secondary amines. In addition, solvent leakage and corrosion are also a problem. So an alternative is to use the various amine-functionalized materials. Adsorption of pollutant gases like CO2 can be done by these functionalized materials. Zhang et al.155 studied the adsorption properties of CO2 on MCM-41 mesoporous materials impregnated with ethylenediamine (EDA), tetraethylenepentamine (TEPA) and two kinds of polyethylene imines (PEI600 and PEI1800). According to the study, the EDA-impregnated samples showed a low CO2 adsorption capacity due to volatilization of EDA compared to the TEPA-impregnated ones. The adsorption capacity decreased with an increase in molecular weight of the amines. A maximum CO2 adsorption capacity of 2.7 mmol g−1 was achieved on TEPA-MCM-41 with 40% TEPA. The work of Kamarudin et al.156 discusses CO2 adsorption on solid adsorbent in a pressure swing adsorption system and its regeneration performance. According to that study, among monoethylamine (MEA)- and diethylamine (DEA)-modified MCM-41, 25 wt% MEA has high CO2 removal even after 10 cycles of operation. Various polyethylene imine (PEI)-modified mesoporous silicas such as MCM-41, MCM-48 and SBA-15 were studied for the CO2 adsorption phenomena by Sharma et al.157 All the PEI-loaded pelletized materials exhibited substantially high reversible CO2 adsorption–desorption behaviors with >99% recovery. The results indicate that pellets containing methyl cellulose and activated carbon show better mechanical strength and CO2 adsorption. That study also proved that MCM-48 is a better material as compared to MCM-41 and SBA-15 for pelletization and loading of PEI. López-Aranguren et al.158 examined the functionalization of silica supports via supercritical CO2 grafting of aminosilanes, which is an important step in the preparation of materials used as solid sorbents in CO2 capture. Four materials have been considered as solid supports: two commercially available silica gels (4.1 and 8.8 nm pore diameter), mesoporous silica MCM-41 (3.8 nm pore diameter) and a microporous faujasite of the Y type. Mono- and diaminotrialkoxysilane were chosen for this study. Through various characterizations it was found that the aminosilane groups were covalently attached to the amorphous silica surface in the mesoporous supports, but not in the microporous zeolite. The amine-functionalized MCM-41 and the 8.8 nm silica gel exhibited a significantly higher uptake of CO2 at low pressures compared with the bare supports. On the contrary, for the 4 nm silica gel and the zeolite the adsorption decreased after impregnation.5.2 Adsorption of heavy metals and organic pollutants
Due to their high surface area, MCM-41 materials can act as excellent adsorbents for heavy metals. The amine-modified samples can easily extract various heavy metals from water by forming covalent bonds with them. Speciation and separation of chromium(VI) and chromium(III) from aqueous solutions were investigated by many research groups. Idris et al.159 studied aminopropyl-functionalized mesoporous silica (AP-MCM-41) as an adsorbent for Cr(VI) and Cr(III). The as-synthesized adsorbent was produced following a simple synthesis method at room temperature prior to template removal using microwave digestion. AP is a simple chelate, yet it can extract Cr(VI) exclusively from solutions containing other mixed metal ions simply by tuning the solution pH. Recovery of Cr(VI) from loaded AP-MCM-41 is also easy to perform with 100% extraction efficiencies. The materials can be reused several times without losing their activity. The ability of various as-prepared and organically modified MCM-41 and HMM mesoporous silica materials to behave as efficient adsorbents for organic pollutants in aqueous solution was investigated by using different surface functionalization procedures, so as to adjust their hydrophilic/hydrophobic balance. For highly organically functionalized samples, the residual superficial silanol groups (<50%) are sufficiently isolated from each other so as to prevent water capillary condensation within the pores, thereby leading to an increased hydrophobic character of the resulting mesoporous silica. The adsorption and storage capacity of the MCM-41 materials increased by 20 times after amine functionalization towards the organic pollutant N,N-diethyl-m-toluamide.160 Parida et al.161 showed that a 12.8 wt% APTES-modified MCM-41 acted as the best catalyst for the adsorption of Cu from aqueous solution. The adsorbed Cu can again be used in the epoxidation of styrene with good conversion and selectivity.5.3 Catalysis of liquid-phase reactions
The organic amine-modified MCM-41 materials are mainly used as base catalysts in liquid-phase reactions. According to Parida et al.,162 NH2-MCM-41 is an efficient catalyst for the Knoevenagel condensation reaction. The effect of amine group can be studied by varying the percentage of APTES. Again the NH2-MCM-41 material is further modified with Cu metal and utilized in single-step amination of benzene.163 Cu/NH2-MCM-41 with Si/Cu = 20 showed a maximum 72% conversion with 100% selectivity for aniline. Díaz et al.164 studied dialkylsilane-functionalized MCM-41 for the esterification of glycerol with fatty acids such as lauric and oleic acids. According to Choudary et al.,165 diamine-functionalized MCM-41 materials show good activity towards Knoevenagel and Aldol condensation reactions. The covalently bonded amine-modified MCM-41 materials can act as excellent CO2 capture agents. Again, CO2 reacts with 2-aminobenzonitriles to form a wide variety of quinazoline-2,4(1H,3H)-dione derivatives. Further, the intermediates are used for the synthesis of biologically active derivatives such as prazosin, bunazosin and doxazosin.166,167 The amine-functionalized materials can be further modified by various other active groups (i.e. heteropoly acids, transition metals, sulphonic acid etc.) for further catalytic applications.168,1696. Thermal stability of amine-functionalized mesoporous silica
In the industrial application of these materials, thermal stability plays an important role. The surface of mesoporous silica materials is hydrophilic in nature due to the presence of silanol groups. After various amine modifications it becomes hydrophobic as alkyl chains replace the silanol groups. These surface modifications influence the thermal and hydrothermal stability of the functionalized materials. Wei et al.170 discussed the variation of thermal stability with amine modification of SBA-15 and SBA-16 materials. They showed the thermal stability of N-(2-aminoethyl)-3-aminopropyltrimethoxysilane (AEAPS)-modified SBA-16 in both He and air atmosphere. AEAPS-SBA-16 was found to be stable up to 200 °C in He and also in air. Ethylenediamine-SBA-15 was stable up to 300 °C in He and 200 °C in air.171 Varying the amine to APTES, the APTES-SBA-15 material was found to be stable up to 250 °C.172 Huang et al.173 also showed that APTES-MCM-48 was thermally stable up to 200 °C. AEAPS-modified SBA-16 can be used as a good adsorbent for CO2 gas.174 The thermal stability of this material was studied by TG-DTG measurement and found to be stable up to 200 °C. The thermal stability of amine-functionalized materials increased with an increase in molecular weight of the amine, although the adsorption capacity decreased with molecular weight of the amine. Zhi-lin et al.175 studied various amine-modified MCM-41 materials and found that PEI-modified materials are maximally stable up to 175 °C. Hence thermal stability plays a vital role in the applications of these materials.1767. Conclusions
As there are increasing economic, social and environmental demands for safe and green research, the contribution of multi-active amine-modified MCM-41 materials becomes a prominent area of research. Simply mixing homogeneous and heterogeneous catalysts is not the solution for the search for new materials. The harmful effects of catalysts cannot be avoided by the traditional methods. Hence the design of inorganic–organic hybrid materials can be an important solution to deal with these challenges.In this review the synthesis and modification of different hybrid materials, especially amine-modified mesoporous silica materials, and the impact of these modifications on their catalytic activities are discussed through various examples. The sole aim of this review is to attract the attention of more and more researchers towards this versatile material. Also some new and interesting applications can be designed with further innovative research in this field.
Notes and references
- M. M. Ardakani, M. A. S. Mohseni, M. A. Alibeik and A. Benvidi, Analyst, 2012, 137(8), 1950 RSC.
- M. Schoeffel, N. Brodie-Linder, F. Audonnetx and C. Alba-Simionesco, J. Mater. Chem., 2012, 22, 557 RSC.
- F. Schuth, K. Sing and J. Weitkamp, Handbook of Porous Solids, Wiley-VCH, Weinheim, 2002, vol. I–V Search PubMed.
- G. A. Ozin and C. Gil, Chem. Rev., 1989, 89, 1749 CrossRef CAS.
- S. L. Gillet, Nanotechnology, 1996, 7, 168 CrossRef.
- T. Yanagisawa, T. Schhimizu, K. Kuroda and C. Kato, Bull. Chem. Soc. Jpn., 1990, 63, 988 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T.-W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenkert, J. Am. Chem. Soc., 1992, 114, 10834 CrossRef CAS.
- B. Trewyn, Acc. Chem. Res., 2007, 40, 846 CrossRef CAS PubMed.
- D. Zhao, Science, 1998, 279, 548 CrossRef CAS.
- P. Selvam, S. K. Bhatia and C. G. Sonwane, Ind. Eng. Chem. Res., 2001, 40, 3237 CrossRef CAS.
- A. B. D. Nandiyanto, F. Iskandar and K. Okuyama, Chem. Lett., 2008, 37, 1040 CrossRef CAS.
- A. B. D. Nandiyanto, S. G. Kim, F. Iskandar and K. Okuyama, Microporous Mesoporous Mater., 2009, 120, 447 CrossRef CAS PubMed.
- A. Corma, Chem. Rev., 1995, 95, 559 CrossRef CAS.
- Q. Huo, D. I. Margolese, U. Ciesla, P. Feng, T. E. Gier, P. Sieger, R. Leon, P. M. Petroff, F. Schuth and G. D. Stucky, Nature, 1994, 368, 317 CrossRef CAS.
- Q. Huo, R. Leon and G. D. Stucky, Science, 1995, 268, 1324 CAS.
- D. Zhao, Q. Huo, J. Feng, J. Kim, Y. Han and G. D. Stucky, Chem. Mater., 1999, 11, 2668 CrossRef CAS.
- D. Zhao, Q. Huo, J. Feng, J. Kim, Y. Han and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024 CrossRef CAS.
- W. W. Lukens Jr, P. Schmidt-Winkel, D. Zhao, J. Feng and G. D. Stucky, Langmuir, 1999, 15, 5403 CrossRef.
- S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 1999, 121, 9611 CrossRef CAS.
- A. Monnier, F. Schuth, Q. Huo, D. Kumar, D. Margolese, R. S. Maxwell, G. Stucky, M. Krishnamurty, P. Petroff, A. Firouzi, M. Janicke and B. Chmelka, Science, 1993, 261, 1299 CAS.
- Q. Huo, D. Margolese, U. Ciesla, P. Feng, T. Gier, P. Sieger, R. Leon, P. M. Petroff, U. Ciesla, F. Schuth and G. Stucky, Nature, 1994, 368, 317 CrossRef CAS.
- Q. Huo, D. Margolese, U. Ciesla, D. Demuth, P. Feng, T. Gier, P. Sieger, A. Firouzi, B. Chmelka, F. Schuth and G. D. Stucky, Chem. Mater., 1994, 6, 1176 CrossRef CAS.
- G. S. Attard, J. C. Glyde and C. G. Göltner, Nature, 1995, 378, 366 CrossRef CAS.
- R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743 CrossRef CAS.
- A. H. Lu, W. Schmidt, A. Taguchi, B. Spliethoff, B. Tesche and F. Schüth, Angew. Chem., Int. Ed., 2002, 41, 3489 CrossRef CAS.
- J. C. Vartuli, K. D. Schmitt, C. T. Kresge, W. J. Roth, M. E. Leonowicz, S. B. McCullen, S. D. Hellring, J. S. Beck, J. L. Schlenker, D. H. Olson and E. W. Sheppard, Chem. Mater., 1994, 6, 2317 CrossRef CAS.
- Z. A. Alothman, Materials, 2012, 5, 2874 CrossRef CAS PubMed.
- A. E. Garcia-Bennett, O. Terasaki, S. Che and T. Tatsumi, Chem. Mater., 2004, 16, 813 CrossRef CAS.
- M. Linden, P. Agren, S. Karlsson, P. Bussian and H. Amenitsch, Langmuir, 2000, 16, 5831 CrossRef CAS.
- N. Ulagappan and C. N. R. Rao, Chem. Commun., 1996, 2759 RSC.
- A. Lind, J. Andersson, S. Karlsson, P. Agren, P. Bussian, H. Amenitsch and M. Linden, Langmuir, 2002, 18, 1380 CrossRef CAS.
- S. Namba, A. Mochizuki and M. Kito, Chem. Lett., 1998, 7, 569 CrossRef.
- G. S. Attard, J. C. Glyde and C. G. Goltner, Nature, 1995, 378, 366 CrossRef CAS.
- D. Khushalani, G. A. Ozin and A. Kuperman, J. Mater. Chem., 1999, 9, 1491 RSC.
- O. Franke, J. Rathousky, G. Schulz-Ekloff and A. Zukal, Stud. Surf. Sci. Catal., 1995, 91, 309 CrossRef CAS.
- F. P. Matthae, D. Genske, C. Minchev and H. Lechert, Stud. Surf. Sci. Catal., 1998, 117, 223 CrossRef CAS.
- Y. R. Cheng, H. P. Lin and C. Y. Mou, Langmuir, 1998, 10, 3772 Search PubMed.
- R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743 CrossRef CAS.
- A. H. Lu, W. Schmidt, A. Taguchi, B. Spliethoff, B. Tesche and F. Schüth, Angew. Chem., Int. Ed., 2002, 41, 3489 CrossRef CAS.
- Q. Huo, D. I. Margolese, U. Ciesla, P. Feng, T. E. Gier, P. Sieger, R. Leon, P. M. Petroff, F. SchEth and G. D. Stucky, Nature, 1994, 368, 317 CrossRef CAS.
- Q. Huo, D. I. Margolese, U. Ciesla, D. G. Demuth, P. Feng, T. E. Gier, P. Sieger, A. Firouzi, B. F. Chmelka, F. SchEth and G. D. Stucky, Chem. Mater., 1994, 6, 1176 CrossRef CAS.
- J. C. Vartuli, K. D. Schmitt, C. T. Kresge, W. J. Roth, M. E. Leonowicz, S. B. McCullen, S. D. Hellring, J. S. Beck, J. L. Schlenker, D. H. Olson and E. W. Sheppard, Chem. Mater., 1994, 6, 2317 CrossRef CAS.
- S. Che, A. E. Garcia-Bennett, T. Yokoi, K. Sakamoto, H. Kunieda, O. Terasaki and T. Tatsumi, Nat. Mater., 2003, 2, 801 CrossRef CAS PubMed.
- A. E. Garcia-Bennett, O. Terasaki, S. Che and T. Tatsumi, Chem. Mater., 2004, 16, 813 CrossRef CAS.
- M. Linden, P. Agren, S. Karlsson, P. Bussian and H. Amenitsch, Langmuir, 2000, 16, 5831 CrossRef CAS.
- N. Ulagappan and C. N. R. Rao, Chem. Commun., 1996, 2759 RSC.
- A. Lind, J. Andersson, S. Karlsson, P. Agren, P. Bussian, H. Amenitsch and M. Linden, Langmuir, 2002, 18, 1380 CrossRef CAS.
- N. K. Raman, M. T. Anderson and C. J. Brinker, Chem. Mater., 1996, 8, 1682 CrossRef CAS.
- O. Franke, J. Rathousky, G. Schulz-Ekloff and A. Zukal, Stud. Surf. Sci. Catal., 1995, 91, 309 CrossRef CAS.
- F. P. Matthae, D. Genske, C. Minchev and H. Lechert, Stud. Surf. Sci. Catal., 1998, 117, 223 CrossRef CAS.
- H. P. Lin, Y. R. Cheng and C. Y. Mou, Chem. Mater., 1998, 10, 3772 CrossRef CAS.
- S. Namba, A. Mochizuki and M. Kito, Chem. Lett., 1998, 7, 569 CrossRef.
- R. Mokaya, W. Jones, Z. Luan, M. D. Alba and J. Kilinowski, Catal. Lett., 1996, 37, 113 CrossRef CAS.
- M. Selvaraj, P. K. Sinha and A. Pandurangan, Microporous Mesoporous Mater., 2004, 70, 81 CrossRef CAS PubMed.
- M. Selvaraj, A. Panurangan, K. S. Seshadri, P. K. Sinha, V. Krishnasamy and K. B. Lal, Appl. Catal., A, 2003, 242, 347 CrossRef CAS.
- M. Selvaraj, B. R. Min, Y. G. Shul and T. G. Lee, Microporous Mesoporous Mater., 2004, 74, 143 CAS.
- W. C. Li, Y. C. Chih and N. K. Na, Appl. Catal., A, 1998, 178, 1 CrossRef.
- J. M. F. B. Aquino, C. D. R. Souza and A. S. Araujo, Int. J. Inorg. Mater., 2001, 3, 467 CrossRef CAS.
- L. T. Zhuravlev, Langmuir, 1987, 3, 316 CrossRef CAS.
- X. S. Zhao, G. Q. Lu, A. K. Whittaker, G. J. Millar and H. Y. Zhu, J. Phys. Chem. B, 1997, 101, 6525 CrossRef CAS.
- H. Landmesser, H. Kosslick, W. Storek and R. Fricke, Solid State Ionics, 1997, 101, 271 CrossRef.
- M. F. Ottaviani, A. Galarneau, D. Desplantiers-Giscard, F. Di Renzo and F. Fajula, Microporous Mesoporous Mater., 2001, 44, 1 CrossRef.
- A. Galarneau, D. Desplantier-Giscard, F. di Renzo and F. Fajula, Catal. Today, 2001, 68, 191 CrossRef CAS.
- M. E. Davis, Nature, 2002, 417, 813 CrossRef CAS PubMed.
- A. Katovic, G. Giordano, B. Bonelli, B. Onida, E. Garrone, P. Lentz and J. B. Nagy, Microporous Mesoporous Mater., 2001, 44, 275 CrossRef.
- T. Blasco, A. Corma, M. T. Navarro and J. P. Pariente, J. Catal., 1995, 156, 65 CrossRef CAS.
- D. Wei, H. Wang, X. Feng, W. T. Chueb, P. Ravikovitch, M. Lyubovsky, C. Li, T. Takeguchi and G. L. Haller, J. Phys. Chem. B, 1999, 103, 2113 CrossRef CAS.
- A. Sayari, C. Danumah and I. L. Moudrakovshi, Chem. Mater., 1995, 7, 813 CrossRef CAS.
- M. A. Karakassides, K. G. Fournaris, A. Travlos and D. Petridis, Adv. Mater., 1998, 10, 483 CrossRef CAS.
- Z. Chang, Z. Zhu and K. Larry, J. Phys. Chem. B, 1999, 103, 9442 CrossRef CAS.
- Z. Chang, J. Suo, X. Zhang and S. Li, Chem. Commun., 1998, 8, 241 Search PubMed.
- L. Wang, J. Shi, J. Yu and D. Yan, Nanostruct. Mater., 1998, 10, 1289 CrossRef CAS.
- A. Corma, Chem. Rev., 1997, 97, 2373 CrossRef CAS PubMed.
- A. Corma and D. Kumar, Stud. Surf. Sci. Catal., 1998, 117, 201 CrossRef CAS.
- A. Sayari, Chem. Mater., 1996, 8, 1840 CrossRef CAS.
- A. Corma, M. T. Navarro and J. Pérez-Pariente, Chem. Commun., 1994, 147 RSC.
- P. T. Tanev, M. Chibwe and T. J. Pinnavaia, Nature, 1994, 368, 321 CrossRef CAS PubMed.
- R. J. Mahalingam, S. K. Badamali and P. Selvam, Chem. Lett., 1999, 11, 1141 CrossRef.
- F. Schuth, Phys. Chem., 1995, 99, 1306 CrossRef.
- I. V. Kozhevnikov, A. Sinnema, R. J. J. Jansen, K. Pamin and H. van Bekkum, Catal. Lett., 1995, 30, 241 CrossRef.
- P. Selvam, S. K. Badamali, M. Murugesan and H. Kuwano, Recent Trends in Catalysis, 1999, p. 545 Search PubMed.
- R. Long and R. T. Yang, Catal. Lett., 1998, 52, 91 CrossRef CAS.
- U. Junges, W. Jacobs, I. Giogt-Martin, B. Krutzsch and F. Schuth, Chem. Commun., 1995, 2283 RSC.
- A. Corma, A. Martinez, V. Martinez-Soria and J. B. Monton, J. Catal., 1995, 153, 25 CrossRef CAS.
- F. M. T. Mendes and M. Schmal, Appl. Catal., A, 1997, 163, 153 CrossRef CAS.
- H. F. Chang and M. L. Saleque, J. Mol. Catal. A: Chem., 1994, 88, 223 CrossRef CAS.
- V. Z. Fridman and A. A. Davydov, J. Catal., 2000, 195, 20 CrossRef CAS.
- J. Becker, J. P. M. Niederer, M. Keller and W. F. Holderich, Appl. Catal., A, 2000, 197, 229 CrossRef CAS.
- R. Q. Long and R. T. Yang, Ind. Eng. Chem. Res., 1999, 38, 873 CrossRef CAS.
- J. Feng, X. Hu, P. L. Yue, H. Y. Zhu and G. Q. Lu, Chem. Eng. Sci., 2003, 58, 679 CrossRef CAS.
- F. Pinna, Catal. Today, 1998, 41, 129 CrossRef CAS.
- X. Hu, F. L. Y. Lam, L. M. Cheung, K. F. Chan, X. S. Zhao and G. Q. Lu, Catal. Today, 2001, 68, 129 CrossRef CAS.
- C. Dossi, R. Psaro, A. Bartsch, E. Brivio, A. Galasco and P. Losi, Catal. Today, 1993, 17, 527 CrossRef CAS.
- H. Kanzaki, T. Kitamura, R. Hamada, S. Nishiyama and S. Tsuruya, J. Mol. Catal. A: Chem., 2004, 208, 203 CrossRef CAS.
- T. Ohtani, S. Nishiyama, S. Tsuruya and M. Masai, in Proceedings of 10th International Congress on Catalysis, Budapest, 1992, p. 1999 Search PubMed.
- J. Okamura, S. Nishiyama, S. Tsuruya, M. Masai and M. Masai, J. Mol. Catal. A: Chem., 1998, 135, 133 CrossRef CAS.
- T. Ohtani, S. Nishiyama, S. Tsuruya and M. Masai, J. Catal., 1995, 155, 158 CrossRef CAS.
- E. Antonakou, A. Lappas, M. H. Nilsen, A. Bouzga and M. Stocker, Fuel, 2006, 85, 2202 CrossRef CAS PubMed.
- A. Tuel, Microporous Mesoporous Mater., 1999, 27, 151 CrossRef CAS.
- J. Xin, J. Suo and X. Zhang, New J. Chem., 2000, 24, 569 RSC.
- A. Corma, Chem. Rev., 1997, 97, 2373 CrossRef CAS PubMed.
- V. Umamaheshwari, M. Palanichamy and V. Murugesan, J. Catal., 2002, 210, 367 CrossRef.
- A. Wingen, N. Anastasievie, A. Hollnagel, D. Werner and F. Schuth, J. Catal., 2000, 193, 248 CrossRef CAS.
- V. Parvulescu and B. L. Su, Catal. Today, 2002, 69, 315 CrossRef.
- K. M. Reddy, B. Wei and C. Song, Catal. Today, 1998, 43, 261 CrossRef CAS.
- V. Parvulescu, C. Constantin and B. L. Su, J. Mol. Catal. A: Chem., 2003, 202, 171 CrossRef CAS.
- V. Parvulescu, C. Dascalescu and B. L. Su, Stud. Surf. Sci. Catal., 2001, 135, 4772 CrossRef CAS.
- V. Parvulescu, C. Anastasescu, C. Constantin and B. L. Su, Stud. Surf. Sci. Catal., 2002, 142, 1204 Search PubMed.
- B. Coq and F. Figueras, J. Mol. Catal. A: Chem., 2001, 173, 117 CrossRef CAS.
- N. Savargaonkar, B. C. Khanra, M. Pruski and T. S. King, J. Catal., 1996, 162, 277 CrossRef CAS.
- V. Pârvulescu, Cr. Tablet, C. Anastasescu and B. L. Su, J. Mol. Catal. A: Chem., 2004, 211, 165 CrossRef PubMed.
- S. Velu, L. Wang, M. Okazaki, K. Suzuki and S. Tomura, Microporous Mesoporous Mater., 2002, 54, 113 CrossRef CAS.
- R. Savidha, A. Pandurangan, M. Palanichamy and V. Murugesan, J. Mol. Catal. A: Chem., 2004, 211, 165 CrossRef CAS PubMed.
- Y. Kubota, Y. Nishizaki and Y. Sugi, Chem. Lett., 2000, 29, 998 CrossRef.
- J. Wu, X. Liu and S. H. Tolbert, J. Phys. Chem. B, 2000, 104, 11837 CrossRef CAS.
- J. Wu, M. M. Abu-Omar and S. H. Tolbert, Nanoletters, 2001, 1, 27 CrossRef CAS.
- J. F. Diaz, J. J. Balkus, F. Bedioui, V. Kurshev and L. Kevan, Chem. Mater., 1997, 9, 61 CrossRef CAS.
- L. Mercier and T. J. Pinnavaia, Adv. Mater., 1997, 9, 500 CrossRef CAS.
- S. L. Burkett, S. D. Sims and S. Mann, Chem. Commun., 1996, 1367 RSC.
- M. H. Lim, C. F. Blanford and A. Stein, J. Am. Chem. Soc., 1997, 119, 4090 CrossRef CAS.
- V. Antochshuk, A. S. Araujo and M. Jaroniec, J. Phys. Chem. B, 2000, 104, 9713 CrossRef CAS.
- C. Yoshina-Ishii, T. Asefa, N. Coombs, M. J. MacLachlan and G. A. Ozin, Chem. Commun., 1999, 2539 RSC.
- T. Asefa, M. J. MacLachlan, N. Coombs and G. A. Ozin, Nature, 1999, 402, 867 CAS.
- S. Inagaki, S. Guan, Y. Fukushima, T. Oshuma and O. Terasaki, J. Am. Chem. Soc., 1999, 121, 9611 CrossRef CAS.
- B. J. Melde, B. T. Holland, C. F. Blanford and A. Stein, Chem. Mater., 1999, 11, 3302 CrossRef CAS.
- T. Asefa, C. Yoshina-Ishii, M. J. MacLachlan and G. A. Ozin, J. Mater. Chem., 2000, 10, 1751 RSC.
- A. B. Bourlinos, Th. Karakostas and D. Petridis, J. Phys. Chem. B, 2003, 107, 920 CrossRef CAS.
- K. J. Shea and D. A. Loy, Chem. Mater., 2001, 13, 3306 CrossRef CAS.
- F. Hoffmann, M. Cornelius, J. Morell and M. Froba, Angew. Chem., Int. Ed., 2006, 45, 3216 CrossRef CAS PubMed.
- C. N. Perez, E. Moreno, C. A. Henriques, S. Valange, Z. Gabelica and J. L. F. Monteirio, Microporous Mesoporous Mater., 2000, 48, 137 CrossRef.
- C. M. Yang and K. J. Chao, J. Chin. Chem. Soc., 2002, 49, 883 CAS.
- Y. Kubota, Y. Nishizaki, H. Ikeya, M. Saeki, T. Hida, S. Kawazu, M. Yoshida, H. Fuji and Y. Sugi, Microporous Mesoporous Mater., 2004, 70, 135 CrossRef CAS PubMed.
- Y. Kubota, H. Ikeya, Y. Sugi, T. Yamada and T. Tatsumi, J. Mol. Catal. A: Chem., 2006, 249, 181 CrossRef CAS PubMed.
- L. Martins, T. J. Bonagama, E. R. De Azevedo, P. Bargiela and D. Cardoso, Appl. Catal., A, 2006, 312, 77 CrossRef CAS PubMed.
- L. Martins and D. Cardoso, Microporous Mesoporous Mater., 2006, 106, 8 CrossRef PubMed.
- S. Huh, J. W. Wiench, J. Yoo, M. Prusky and V. S. Y. Lin, Chem. Mater., 2003, 15, 4247 CrossRef CAS.
- H. I. Meléndez-Ortiz, Y. P. Mercado, J. A. Mercado-Silva, Y. Olivares-Maldonado, G. Castruita and L. A. García-Cerda, Ceram. Int., 2014, 40, 9701 CrossRef PubMed.
- M. Gil, I. Tiscornia, Ó. de la Iglesia, R. Mallada and J. Santamaría, Chem. Eng. J., 2011, 175, 291 CrossRef CAS PubMed.
- X. Yan, S. Komarneni and Z. Yan, J. Colloid Interface Sci., 2013, 390, 217 CrossRef CAS PubMed.
- E. Da'na and A. Sayari, Chem. Eng. J., 2011, 167, 91 CrossRef PubMed.
- E. Da'na and A. Sayari, Desalination, 2012, 285, 62 CrossRef PubMed.
- P. N. Diagboya, B. I. Olu-Owolabi and K. O. Adebowale, J. Environ. Manage., 2014, 146, 42 CrossRef CAS PubMed.
- A. Nomura and C. W. Jones, ACS Appl. Mater. Interfaces, 2013, 5, 5569 CAS.
- B. Levasseur, A. M. Ebrahim and T. J. Bandosz, Langmuir, 2012, 28, 5703 CrossRef CAS PubMed.
- P. Gao, T. Zhang, Y. Wang, C. Gao and Y. Zhao, Mater. Lett., 2011, 65, 260 CrossRef CAS PubMed.
- S. K. Samantaray and K. M. Parida, Catal. Commun., 2005, 6, 578 CrossRef CAS PubMed.
- M. Sahu, S. Singha and K. M. Parida, New J. Chem., 2011, 35, 2503 RSC.
- K. M. Parida, S. Mallick, P. C. Sahoo and S. K. Rana, Appl. Catal., A, 2010, 381, 226 CrossRef CAS PubMed.
- S. Rana, S. Mallick and K. M. Parida, Ind. Eng. Chem. Res., 2011, 50, 2055 CrossRef CAS.
- S. Mallick, S. Rana and K. M. Parida, Dalton Trans., 2011, 40, 9169 RSC.
- G. B. B. Varadwaj, S. Rana and K. M. Parida, RSC Adv., 2013, 3, 7570 RSC.
- G. B. B. Varadwaj, S. Rana and K. M. Parida, Dalton Trans., 2013, 42, 5122 RSC.
- F. Song, Y. Zhao and Q. Zhong, J. Environ. Sci., 2013, 25, 554 CrossRef CAS.
- F. Song, Y. Zhao, Y. Cao, J. Ding, Y. Bu and Q. Zhong, Appl. Surf. Sci., 2013, 268, 124 CrossRef CAS PubMed.
- Z. Liu, Y. Teng, K. Zhang, Y. Cao and W. Pan, J. Fuel Chem. Technol., 2013, 41, 469 CrossRef CAS.
- K. S. N. Kamarudin and N. Alias, Fuel Process. Technol., 2013, 106, 332 CrossRef CAS PubMed.
- P. Sharma, J. Seong, Y. Jung, S. Choi, S. Park, Y. Yoon II and I.-H. Baek, Powder Technol., 2012, 219, 86 CrossRef CAS PubMed.
- P. López-Aranguren, J. Fraile, L. F. Vega and C. Domingo, J. Supercrit. Fluids, 2014, 85, 68 CrossRef PubMed.
- S. A. Idris, K. Alotaibi, T. A. Peshkur, P. Anderson and L. T. Gibson, J. Colloid Interface Sci., 2012, 386, 344 CrossRef CAS PubMed.
- A. Trouvé, I. Batonneau-Gener, S. Valange, M. Bonne and S. Mignard, J. Hazard. Mater., 2012, 201, 107 CrossRef PubMed.
- K. M. Parida, K. G. Mishra and S. K. Dash, Ind. Eng. Chem. Res., 2012, 51, 2235 CrossRef CAS.
- K. M. Parida and D. Rath, J. Mol. Catal. A: Chem., 2009, 310, 93 CrossRef CAS PubMed.
- K. M. Parida, D. Rath and S. S. Dash, J. Mol. Catal. A: Chem., 2010, 318, 85 CrossRef CAS PubMed.
- I. Díaz, F. Mohino, J. Pérez-Pariente and E. Sastre, Appl. Catal., A, 2003, 242, 161 CrossRef.
- B. M. Choudary, M. Lakshmi Kantam, P. Sreekanth, T. Bandopadhyay, F. Figueras and A. Tuel, J. Mol. Catal. A: Chem., 1999, 142, 361 CrossRef CAS.
- D. B. Nale, S. K. Rana, K. M. Parida and B. M. Bhanage, Catal.: Sci. Technol., 2014, 4, 1608 CAS.
- D. B. Nale, S. K. Rana, K. M. Parida and B. M. Bhanage, Appl. Catal., A, 2014, 469, 340 CrossRef CAS PubMed.
- X. Dong, D. Wang, K. Li, Y. Zhen, H. Hua and G. Xue, Mater. Res. Bull., 2014, 57, 210 CrossRef CAS PubMed.
- G. Luo, L. Kang, M. Zhua and B. Dai, Fuel Process. Technol., 2014, 118, 20 CrossRef CAS PubMed.
- J. Wei, J. Shi, H. Pan, Q. Su, J. Zhu and Y. Shi, Microporous Mesoporous Mater., 2009, 117, 596 CrossRef CAS PubMed.
- F. Zheng, D. N. Tran, B. J. Busche, G. E. Fryxell, R. S. Addleman, T. S. Zemanian and C. L. Aardahl, Ind. Eng. Chem. Res., 2005, 44, 3099 CrossRef CAS.
- R. A. Khatri, S. S. C. Chuang, Y. Soong and M. Gray, Energy Fuels, 2006, 20, 1514 CrossRef CAS.
- H. Y. Huang, R. T. Yang, D. Chinn and C. L. Munson, Ind. Eng. Chem. Res., 2003, 42, 2427 CrossRef CAS.
- J. Wei, J. Shi, H. Pan, W. Zhao, Q. Ye and Y. Shi, Microporous Mesoporous Mater., 2008, 116, 394 CrossRef CAS PubMed.
- L. Zhi-lin, T. Yang, Z. Kai, C. Yan and P. Wei-ping, J. Fuel Chem. Technol., 2013, 41, 469 CrossRef.
- G. Luo, L. Kang, M. Zhua and B. Dai, Fuel Process. Technol., 2014, 118, 20 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |