
Poly(N-isopropylacrylamide) hydrogels fabricated via click chemistry: well-defined α,ω-bis propargyl linear poly(N-isopropylacrylamide)s as crosslinkers†
Abstract
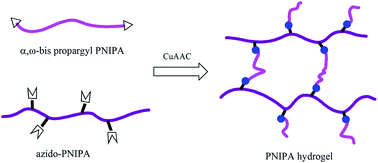
A series of poly(N-isopropylacrylamide) (PNIPA) hydrogels were fabricated through click chemistry by using a well-defined azido-PNIPA carrying pendant azido groups, and linear α,ω-bis propargyl PNIPAs with different chain lengths. Here linear α,ω-bis propargyl PNIPAs were synthesized via reversible addition-fragmentation chain transfer (RAFT) polymerization by using a bis propargyl terminal chain transfer agent, whose chain lengths were modulated by changing polymerization conditions. The obtained hydrogels showed increasing ESR values in the swollen state and increasing volume phase transition temperatures (VPTTs) with increasing molecular weights of bis propargyl PNIPAs, ascribed to the lengthening distance between crosslinks and improving hydrophilicity. The incorporation of amine together with crosslinking modified the hydrophilicity of a click hydrogel, resulting in the elevated VPTT and additional pH sensitivity. The present study provided a facile method to regulate swelling properties and/or to impart special functions for PNIPA hydrogels, by adjusting the chain length of crosslinkers or by introducing other functional groups.
 Please wait while we load your content...
Please wait while we load your content...

