
Photocatalytic NOx abatement: why the selectivity matters†
Abstract
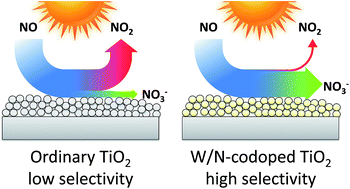
Titanium dioxide photocatalysis offers an excellent way to oxidise NOx to nitrate and thus reduce air pollution. However, unmodified titanium dioxide also releases a significant amount of the toxic intermediate nitrogen dioxide in the process, a problem that is rarely discussed in previous literature. Herein, we highlight this issue by presenting systematic data on the activity and selectivity of a number of commercial titania powders. The photocatalytic performance of a previously developed W/N-codoped titanium dioxide is also reported which, for the first time, offers a way to eliminate this problem as it exhibits an exceptionally high selectivity towards nitrate. The selectivity appears to be solely dependent on the tungsten content, a concentration of 4.8 at.% is sufficient to induce a very high selectivity. Furthermore, the high selectivity could also be replicated by a W/N-codoped sample derived from the industrial sulphate synthetic process. The increased selectivity comes at the expense of absolute activity, which is lower than in the reference titania samples. This raises the question of how to properly evaluate NOx abatement photocatalysts when there are two factors to consider, activity and selectivity. To resolve this, we propose to define a new figure of merit for the evaluation of NOx abatement photocatalysts by distilling total NOx removal and selectivity into one value, the DeNOx index. It is derived by assigning a toxicity value to both NO and NO2 and then expressing the change in total toxicity rather than the concentration change of the individual nitrogen oxides.
 Please wait while we load your content...
Please wait while we load your content...

