
Regioselective palladium-catalysed cross-coupling reactions: a powerful synthetic tool
Abstract
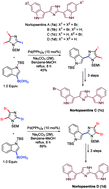
The purpose of this review is to highlight the powerful nature of palladium-catalysed regioselective (site-selective) cross-coupling reactions for facilitating the synthesis of biologically important natural products as well as certain industrially relevant drugs. A total of 28 natural products have been included to highlight the potential of this methodology.
 Please wait while we load your content...
Please wait while we load your content...

