
Chemodosimeter for fluoride ions based on F−-triggered Si–O cleavage followed by the deprotonation/autoxidation of secondary nitrile C–H group†
Abstract
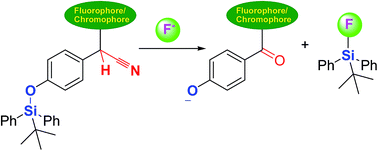
A completely new strategy for a chemodosimeter for fluoride based on F−-triggered Si–O bond cleavage, followed by the deprotonation/autoxidation of secondary nitrile C–H group was developed for the first time. The chemodosimeter exhibited high selectivity and sensitivity, dramatic color “turn-on”, and fluorescence “turn-off” for the detection for fluoride.
 Please wait while we load your content...
Please wait while we load your content...

