
Facile synthesis of PVP-assisted PtRu/RGO nanocomposites with high electrocatalytic performance for methanol oxidation†
Duan Bina,
Fangfang Rena,
Huiwen Wanga,
Ke Zhanga,
Beibei Yanga,
Chunyang Zhaia,
Mingshan Zhu*ab,
Ping Yanga and
Yukou Du*a
aCollege of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, P. R. China. E-mail: duyk@suda.edu.cn; Fax: +86 512 65880089; Tel: +86 512 65880361
bInstitute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: mingshanzhu@yahoo.com
First published on 11th August 2014
Abstract
In this paper, we report a facile approach for the synthesis of polyvinylpyrrolidone (PVP)-stabilized PtRu/RGO nanocomposites (PtRu/RGO/PVP) by the one-pot method. The structure, morphology and composition of the as-prepared catalysts were characterized by Raman, transmission electron microscopy (TEM) and energy dispersive X-ray spectroscopy (EDX), respectively. It was found that PVP plays an important role in controlling the size of PtRu nanoparticles (NPs) as well as their dispersion stability. TEM images show that as-prepared PtRu NPs with a mean particle size of about 3.09 nm are uniformly dispersed on the RGO surface in the presence of PVP. The electrocatalytic properties of the as-prepared catalysts were evaluated by cyclic voltammetry (CV) and chronoamperometry (CA). Compared to PtRu/RGO and PtRu/PVP catalysts, our PtRu/RGO/PVP hybrids exhibited enhanced electrocatalytic activity and stability for the methanol oxidation reaction. Moreover, our multicomposites also showed higher electrocatalytic performance than the commercial PtRu/C catalysts. The PtRu/RGO/PVP nanostructures with an optimized molar ratio of Pt/Ru (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) displayed 1.96 times greater stability than the commercial PtRu/C nanospecies. These findings indicated that PtRu/RGO catalysts show a promising future of potential applications in direct methanol fuel cells with the assistance of PVP stabilized.
:1) displayed 1.96 times greater stability than the commercial PtRu/C nanospecies. These findings indicated that PtRu/RGO catalysts show a promising future of potential applications in direct methanol fuel cells with the assistance of PVP stabilized.
1. Introduction
Due to the continuous consumption of fossil fuel and increasing environment pollution caused by traditional fuel utilization, direct methanol fuel cells (DMFCs) have recently drawn considerable attention and interest as portable devices with the characteristics of high energy density under conditions of low operating temperature and pollutant emission.1–6 At present, pure Pt is recognised as the most active anodic catalyst due to its excellent performance during methanol oxidation.2–5 Unfortunately, the high price of pure Pt and catalyst poisoning by CO-like intermediates during the oxidation of methanol hinder the broad commercialization of DMFCs.2–5Great effort has been focused on both reducing the usage of noble-metal Pt nanoparticles (NPs) and increasing electrocatalytic performance towards methanol oxidation.2–6 One widely accepted strategy is to fabricate a Pt-based binary catalyst by employing a second metal such as metallic Ru. Many studies have demonstrated that the catalytic activity and stability of Pt–Ru catalyst could be evidently enhanced, which could be ascribed to the bifunctional mechanism or electronic effect.4,5,7–16 Another strategy is to adopt catalyst supports to increase the electrochemically active surface area (ECSA) and provide good dispersion for supporting the metal NPs. Various carbon nanomaterials such as carbon black, carbon nanotube (CNT), graphene and their derivatives have served as catalyst supports both academically and commercially.6 Among these supports, graphene has recently received increased attention and has been recognised as a promising candidate for catalyst support due to its extremely high specific surface area, excellent electrical conductivity and superior chemical stability.19,20 These outstanding performances convinced us to consider graphene as an ideal substitute for other carbon materials in fuel cells.
To date, several graphene-hybridized Pt–Ru nanocomposites have been developed for improving electrocatalytic performance.19,20 For example, Dong et al. first reported PtRu nanoparticles of 10 nm supported on graphene with high electrocatalytic performance for methanol and ethanol oxidation.8 Kim and co-authors have reported efficient electrooxidation of glycerol over a PtRu/graphene catalyst.11 Zhao et al. synthesized highly dispersed PtRu/graphene catalysts, which were prepared via the supercritical carbon dioxide–methanol route.13 Liu's group have reported that uniformly distributed PtRu NPs were decorated on the N-doped graphene aerogel with porous structures.16 However, it should be pointed out that the synthetic condition, size and dispersion of metal NPs and catalytic stability for methanol oxidation are still not sufficiently satisfactory to warrant further applications.
A current focus of research is the use of surfactant as a stabilizer to prevent metal colloids from aggregation and improve deposition of colloids onto the carbon support.17,18 For example, the Hwang group reported that graphite carbon nanofibers hybridized with PtRu NPs as promising catalysts with the assistance of polyvinylpyrrolidone (PVP) surfactant.18 The PVP-stabilized nanocomposites efficiently enhanced electrocatalytic performance toward methanol oxidation. More recently, surfactant-assisted graphene-based catalysts distinctly enhanced catalytic activity and stability, because that surfactant efficiently improved the solubility and aggregation prevention of graphene.21 Herein, we develop a one-pot method that produced well-dispersed PtRu NPs with 3.09 nm size supported on the graphene surface in the presence of PVP surfactant. Compared with the bare PtRu/RGO nanospecies, our PVP-assisted PtRu/RGO NPs were not only well separated at 3.09 nm but also showed good stability (stable for more than 1 year and good re-dispersion). Moreover, this multicomposite displayed distinctly improved electrocatalytic activity and stability for PtRu/RGO, PtRu/PVP and commercial PtRu/C nanostructures. With an optimized molar ratio of Pt/Ru (1:1), the PtRu/RGO/PVP nanostructures showed 1.96 times greater stability than the commercial PtRu/C nanospecies. Our investigation might initiate new opportunities for constructing graphene-based materials with enhanced electrocatalytic performance and stability under surfactant assistance and show a promising future of potential applications in direct methanol fuel cells.
2. Experimental
2.1 Chemicals
Graphite powder (Sinopharm Chemicals Reagent Co., Ltd., China) was used as received. Commercial PtRu/C catalysts (JM 30% PtRu, Hispcc5000, Shanghai Hesen Electric Co., Ltd., China), H2PtCl6·6H2O and RuCl3 (Shanghai Shiyi Chemicals Reagent Co., Ltd., China), NaNO3, KMnO4, H2O2 (30%), CH3OH, H2SO4 (95%), polyvinylpyrrolidone (PVP, K30), NaBH4 and other reagents were all of analytical grade purity. All aqueous solutions were prepared with double distilled water.2.2 Preparation of graphene oxide (GO)
GO was prepared from natural graphite powder by a modified Hummers method.22 First, 1.0 g graphite powder and 0.5 g NaNO3 were added to 24 mL concentrated H2SO4 solution with stirring in an ice bath. Then, 3.0 g KMnO4 were slowly added into the solution and the mixture was stirred at 20 °C for 30 min. Next, the temperature of the solution was increased to 35 °C under vigorous stirring for another 30 min, followed by the slow addition of 46 mL of secondary distilled water. Then the temperature was raised to 98 °C and further stirred for 15 min. The reaction was then terminated by adding 140 mL double distilled water after 10 mL 30% H2O2 solution were added. Finally, the resulting solution was filtered and washed with 5% HCl until sulfate ions could not be detected with BaCl2. The sample was dried in a vacuum at 40 °C for 12 hours to obtain the final product of GO.2.3 Synthesis of PVP-stabilized, PtRu/reduced graphene oxide (PtRu/RGO/PVP) nanocomposites
The PVP-stabilized PtRu/RGO nanocomposites were synthesized by using a one-pot method, with NaBH4 as the reducing agent. The preparation process follows: 5 mL of 4.0 mg mL−1 GO and 0.01 g of PVP were dissolved in 20 mL of water accompanied by vigorous stirring for 30 minutes. Then an aqueous solution of 4 mL H2PtCl6 (7.723 × 10−3 mol L−1) and 1.6 mL RuCl3 (19.2 × 10−3 mol L−1), with the molar ratio for Pt to Ru of 1:1, was added to the above mixture with stirring for 1 h. Subsequently, 5 mL of freshly prepared NaBH4 (6 mg mL−1) aqueous solution were added dropwise under vigorous stirring for another 4 h at ambient temperature. The resulting black turbid liquid was centrifuged and repeatedly washed with deionized water and ethanol to remove excess NaBH4, and was then dried for 8 h in a vacuum at 25 °C to obtain the solid product. Finally, the as-prepared catalyst powders were dispersed in 10 mL deionized water and treated ultrasonically for 30 min to obtain the catalyst ink. The obtained catalyst was denoted as PtRu (1:1)/RGO/PVP. For comparison, the PtRu (1:1)/PVP, PtRu (1:1)/RGO, PtRu (1:2)/RGO/PVP, PtRu (2:1)/RGO/PVP and Pt/RGO/PVP were also prepared by the same procedure. All the catalysts contained a total metal (Pt, Pt + Ru) loading of 31%.
2.4 Materials characterization
Digital pictures of the reaction process were taken. The Raman spectra of these samples were recorded at room temperature on a RM2000 microscopic confocal Raman spectrometer with argon-ion laser at the excitation wavelength of 633 nm. The X-ray diffraction (XRD) measurements were performed on a PANalytical X' Pert PRO MRD system with Cu-Kα radiation (k = 1.54056 Å) operated at 40 kV and 30 mA. The morphologies of these samples were characterized using a TECNAI-G20 electron microscope (TEM) with an accelerating voltage of 200 kV. High-resolution TEM (HRTEM) images were obtained with a JEM-2100F high-resolution transmission electron microscope operating at 200 kV. TEM samples were prepared by placing a drop of the colloid dispersion onto a copper grid covered with a perforated carbon film and then evaporating the solvent. The energy dispersive X-ray (EDX) analysis was conducted with a Horiba EMAX X-act energy-dispersive spectroscope attached to the S-4700 system. All measurements were carried out at room temperature.2.5 Electrochemical measurements
All electrochemical measurements were carried out with a CHI 660B electrochemical workstation. A conventional three-electrode system was used with platinum wire, saturated calomel electrode (SCE) and a glassy carbon electrode (GCE, 3 mm diameter) as the counter, reference and working electrodes, respectively. The working electrode was prepared by dropping 20 μL of the composite catalyst ink onto the surface of GCE and dried at room temperature. In this paper, the amount of total metal in glassy carbon was controlled as 0.25 mg cm−2. For the electrochemical active surface area (ECSA) study, cyclic voltammetry (CV) measurements of the catalysts were conducted in a solution of 0.5 M H2SO4 at a scan rate of 50 mV s−1 for 20 cycles from −0.25 V to 1.0 V. The ECSA value can be calculated by integrating the hydrogen adsorption/desorption area between −0.25 V and 0.1 V. The specific ECSA was derived from the following equation:23QH represents the average charge for hydrogen adsorption and desorption, which was theoretically evaluated under the CV curve by QH = ∫IdE/v (v is the scanning rate), m is the loading amount of metal, 0.21 mC cm−2 is the Pt crystalline-activity surface-area transferred coefficient. The electrochemical activity of the methanol oxidation reaction was measured by linear sweep voltammetry at a scan rate of 50 mV s−1 for 20 runs from −0.2 V to 1.0 V. Chronoamperometry (CA) measurements were conducted at 0.6 V for 1500 s. All solutions were deaerated using a dry nitrogen stream and maintained with a slight overpressure of nitrogen during the whole experiment.

3. Results and discussion
Experimentally, the PVP-stabilized PtRu/RGO composites were synthesized by using a one-pot method, where NaBH4 was the reducing agent. First, when GO as the stabilizer in the presence of NaBH4, the noble metal NPs could be efficiently reduced from the H2PtCl6 and RuCl3 precursor as shown in Fig. 1. At the same time, the GO nanosheets were also reduced to reduced graphene oxide (RGO) when NaBH4 solution was dropped into the mix solution.24 The generated noble metal NPs were aggregated and gradually settled after 24 h. In the presence of PVP, the generated noble metal NPs dispersed well in the aqueous solution even after several months. Generally, PVP was used as a stabilizer in the preparation of noble metal nanoclusters. Accordingly, the PVP could efficiently protect the noble metal NPs from aggregation and improve aqueous dispersion. | ||
| Fig. 1 Digital photos of synthetic process for PtRu/RGO, PtRu/PVP and PtRu/RGO/PVP NPs. | ||
The Raman spectrum is a powerful tool to characterize the structure of graphene materials, and corresponding results for GO sheets and our graphene-based materials are displayed in Fig. 2. All three samples exhibit two prominent peaks at approximately 1350 cm−1 and 1598 cm−1, which are attributed to the D and G bands of graphene sheets, respectively.25,26 The D band is usually related to the breathing mode of k-point phonons of A1g symmetry, while the G band is associated with the E2g vibration mode of sp2 carbon domains.25,26 The G band was broadened and shifted towards a slightly higher wave number from GO to RGO, which was probably caused by stress in solution.27 When the GO is chemically reduced, the conjugated graphene network (sp2 carbon) will be reestablished; the intensity ratio of the D and G bands (D/G) increases further because the size of reestablished graphene network becomes smaller than the original.25,26 The peak intensity ratios (ID/IG) are estimated to be 1.32, 1.28, and 0.97 for the PtRu/RGO/PVP, PtRu/RGO and bare GO samples, respectively. Clearly, the PtRu/RGO/PVP and PtRu/RGO catalysts show a higher ID/IG value than that of GO, which demonstrates the effective reduction of GO. Additionally, the ID/IG value of PtRu/RGO/PVP hybrids did not change obviously with the presence of PVP, indicating that the graphene nanosheet did not greatly reduce the size of in-plane sp2 domains.28
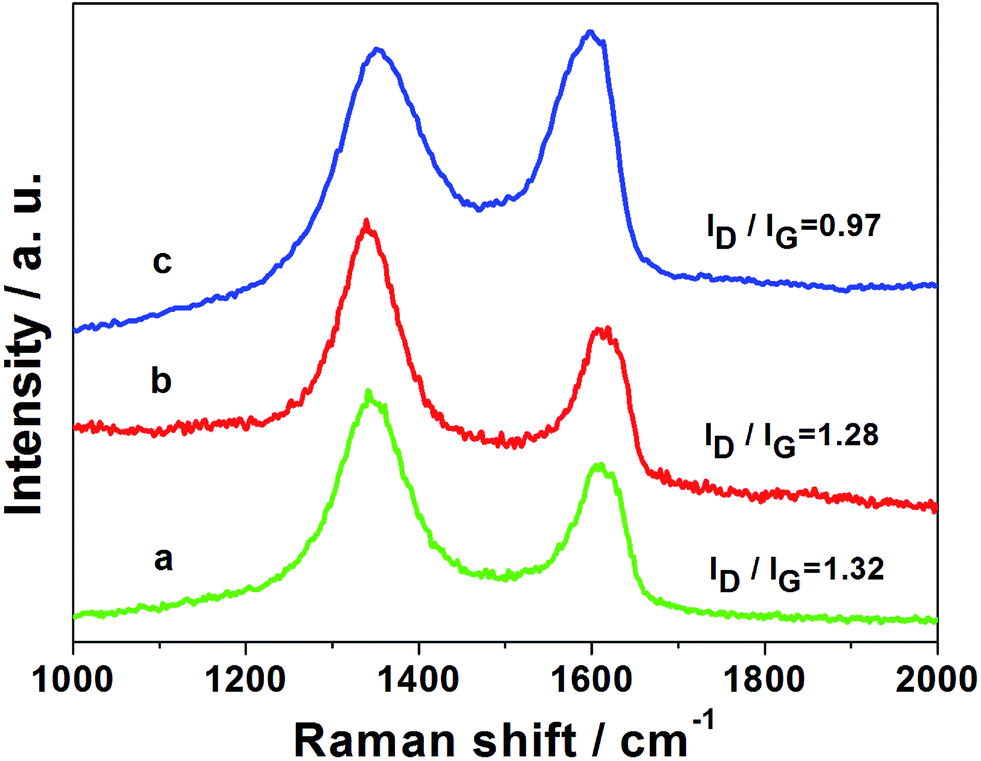 | ||
| Fig. 2 Raman spectra of nanostructures for GO sheets (a), PtRu/RGO (b), and PtRu/RGO/PVP (c). | ||
The morphology and structural features of the PtRu/RGO/PVP, PtRu/RGO and PtRu/PVP catalysts were characterized by TEM analysis. It can be seen from Fig. 3a and b that PtRu NPs are homogeneously distributed on the surface of RGO sheets without obvious agglomeration, suggesting the perfect combination between metal NPs and RGO sheets. The corresponding histogram of particle size distribution in Fig. S1† shows that the PtRu NPs supported on RGO/PVP have a mean diameter of about 3.09 nm. Additionally, the metal NPs in PtRu/PVP composites also present a narrow particle size of about 3.21 nm in Fig. 3c. However, as observed from the TEM image shown in Fig. 3d, PtRu NPs underwent severe aggregation in the PtRu/RGO composites and retained some irregular particles with an average particle size of 5.84 nm, which is much bigger than that of PtRu/RGO/PVP composites. These results illustrate that the addition of PVP helps to prevent PtRu NPs from aggregating and restacking, thereby leading to smaller particle size.29
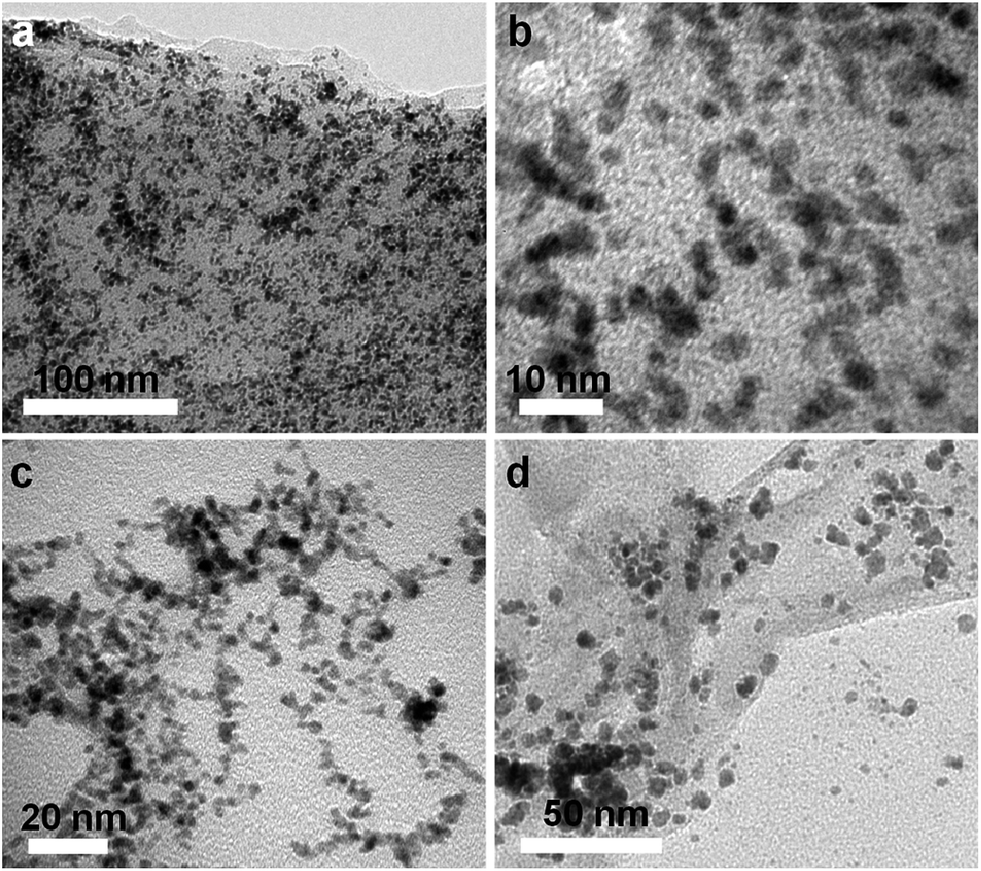 | ||
| Fig. 3 TEM images PtRu/RGO/PVP (a and b), PtRu/PVP (c), and PtRu/RGO (d) nanostructures. | ||
To prove the generation of the PtRu alloy, XRD patterns of Pt/RGO/PVP and various ratios of PtRu/RGO/PVP NPs were investigated. As shown in Fig. 4, for bare Pt/RGO/PVP, the peaks of 39.9°, 46.8° and 67.9° corresponded to diffraction of the (111), (200) and (220) Pt crystal planes (JCPDS no.04-0802), respectively.30 When the Ru atoms were introduced, diffraction peaks shifted slightly to a higher value in the following order: Pt/RGO/PVP, PtRu (2:1)/RGO/PVP, PtRu (1:1)/RGO/PVP and PtRu (1:2)/RGO/PVP (Fig. S2†), which indicated a decrease of the lattice constant as the Ru concentration increased. The position of Pt (111) shifted from 39.9° to 40.8°, which indicated the formation of a bimetallic PtRu alloy in our experiment (Fig. S2†).12,17 Moreover, in addition to the diffraction peaks of Pt or PtRu alloy, the peak at ∼23.2° is characteristic of RGO nanosheets,31 which suggests the presence of graphene in our samples.
 | ||
| Fig. 4 XRD patterns of Pt/RGO/PVP (a), PtRu (2:1)/RGO/PVP (b), PtRu (1:1)/RGO/PVP (c), and PtRu (1:2)/RGO/PVP (d) NPs. | ||
The HRTEM image further reveals that PtRu alloys are crystalline with lattice structures. Generally, the lattice fingers with d-spacing of 0.226 nm can be attributed to the (111) plane of fcc Pt.32 As shown in Fig. 5, the lattice distance for our PtRu/RGO/PVP NPs is measured at 0.22 nm, which is slightly smaller than that of monometallic Pt. Furthermore, the lattice fringes with a lattice spacing of ca. 0.22 nm match with the above (111) PtRu/RGO/PVP plane. This result further indicates the formation of PtRu alloy in our synthetic process. The chemical composition of the PtRu/RGO/PVP composites was analyzed by the EDX spectrum; results are shown in Fig. S3.† The existence of C and O are ascribed to the base plane and oxygenated groups of the RGO, respectively. The spectrum reveals that the atom ratio Pt:Ru is about 1.075, which is very close to the theoretical value (1:1), further suggesting the generated PtRu alloy.
 | ||
| Fig. 5 High-resolution TEM image of PtRu/RGO/PVP NPs. | ||
The electrochemical performances of the PtRu/RGO/PVP, PtRu/RGO and PtRu/PVP catalysts were evaluated by CV analysis in 0.5 M H2SO4 solution. As plotted in Fig. 6 and summarized in Table 1, by integrating the charge in the adsorption/desorption region of hydrogen, we can estimate the value of ECSA at 55.38 m2 g−1 for PtRu/RGO/PVP. Curiously, the PtRu/PVP catalysts showed a negligible ECSA value (3.55 m2 g−1), although the size was similar to that of PtRu/RGO/PVP. When PtRu/RGO nanospecies were used as electrocatalysts, the value of ECSA was estimated at ∼18.70 m2 g−1. This was because the graphene sheets significantly enhanced the ECSA availability of electrocatalysts for electron transfer.33 The larger ECSA of the PtRu/RGO/PVP means that it has more active sites compared to other composites. These results suggested that in the PtRu/RGO/PVP system, the PVP could reduce the size of metal NPs, which increases the density of active sites on the electrode surface, and RGO could improve charge transfer efficiency. The PVP and RGO synergetically contributed to the enhanced electrocatalytic performance. Moreover, our PtRu/RGO/PVP electrode displayed 5 times the ECSA value of commercial PtRu/C species (11.07 m2 g−1), suggesting that the as-prepared electrodes have potential practical fuel cell applications.
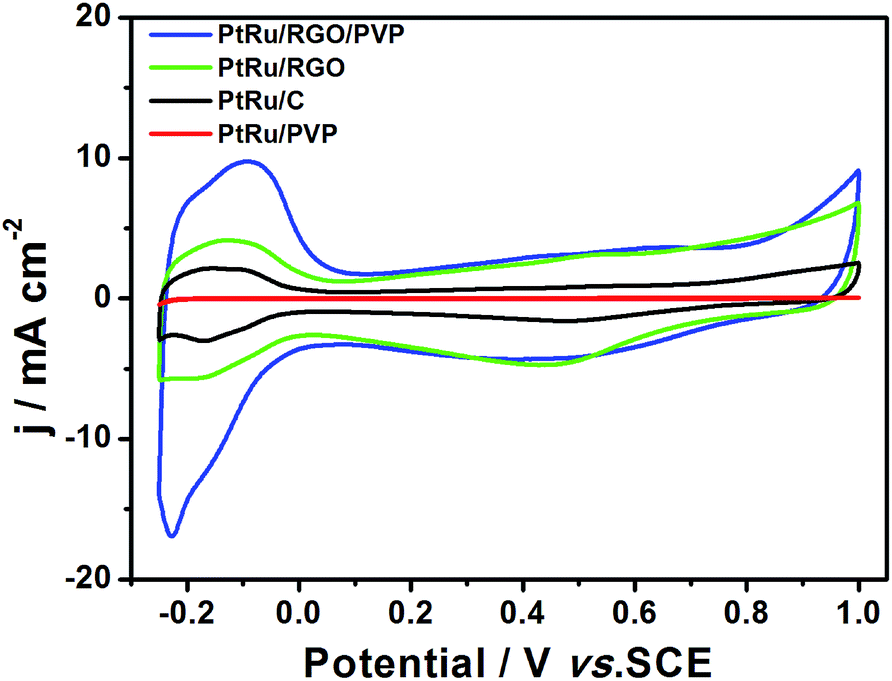 | ||
| Fig. 6 CVs of PtRu/RGO/PVP, PtRu/RGO, PtRu/PVP, and PtRu/C catalysts in 0.5 M H2SO4 solution at scan of 50 mV s−1. | ||
| Electrode | ECSA/m2 g−1 | Forward sweep IF (mA cm−2) (mA mg−1)metal | Reverse sweep Ib (mA cm−2) (mA mg−1)metal | IF/Ib | ||
|---|---|---|---|---|---|---|
| PtRu/RGO/PVP | 55.38 | 36.5 | 146.0 | 19.5 | 78.0 | 1.87 |
| PtRu/RGO | 18.70 | 23.1 | 92.4 | 13.0 | 52.0 | 1.78 |
| PtRu/PVP | 3.55 | 0.2 | 0.8 | 0.18 | 0.72 | 1.11 |
| PtRu/C | 11.07 | 18.6 | 74.4 | 14.2 | 56.8 | 1.31 |
Fig. 7 shows the CVs of methanol oxidation on the PtRu/RGO/PVP, PtRu/RGO, PtRu/PVP and commercial PtRu/C electrodes in 0.5 M H2SO4 containing 1.0 M CH3OH solution. As in Fig. 7 and Table 1, the forward peak current density of methanol oxidation on PtRu/RGO/PVP (36.5 mA cm−2 or 146 mA mg−1) is about 1.6, 29.2 and 1.96 times higher than that on PtRu/RGO (18.48 mA cm−2 or 92.4 mA mg−1), PtRu/PVP (0.2 mA cm−2 or 0.8 mA mg−1) and PtRu/C (18.6 mA cm−2 or 74.4 mA mg−1), respectively. Generally, the ratio of the forward anodic peak current (If) to the backward cathodic peak current (Ib) typically is used for evaluating catalyst tolerance to CO and other carbonaceous species.34 The PtRu/RGO/PVP catalyst has a higher If/Ib (1.87) than the PtRu/RGO (1.78) and commercial PtRu/C (1.31) catalysts. Therefore, based on the above analysis, the as-prepared PtRu/RGO/PVP shows the highest electrocatalytic performance and better tolerance of intermediate species than the other catalysts. This can largely be ascribed to two factors: the improved surface area of PtRu NPs on the graphene sheet support,35 and modification of PVP, which can prevent the PtRu NPs from aggregating in chemical reactions, leading to formation of well-dispersed and smaller-sized PtRu NPs that provided more available active catalytic sites.
 | ||
| Fig. 7 CVs of PtRu/RGO/PVP, PtRu/RGO, PtRu/PVP, PtRu/C catalysts in 0.5 M H2SO4 + 1.0 M CH3OH solution at scan of 50 mV s−1. | ||
The stabilities of the as-prepared and commercial catalysts were also studied. Fig. 8 shows that the CA curves for these catalysts performed in 0.5 M H2SO4 + 1.0 M CH3OH at 0.6 V for 1500 s. In the initial period, a rapid current decay for all catalysts was observed due to the formation of intermediate species such as CH3OHads and CHOads during the methanol oxidation reaction.36 During the whole time, it was clear that the current density produced on the PtRu/RGO/PVP catalyst was higher than the current density produced on the PtRu/RGO, PtRu/C and PtRu/PVP catalysts. This finding indicates that the PtRu/RGO/PVP catalyst has a more durable and higher electrocatalytic activity for methanol oxidation in comparison with the other catalysts, which is also consistent with the CV results (Fig. 7).
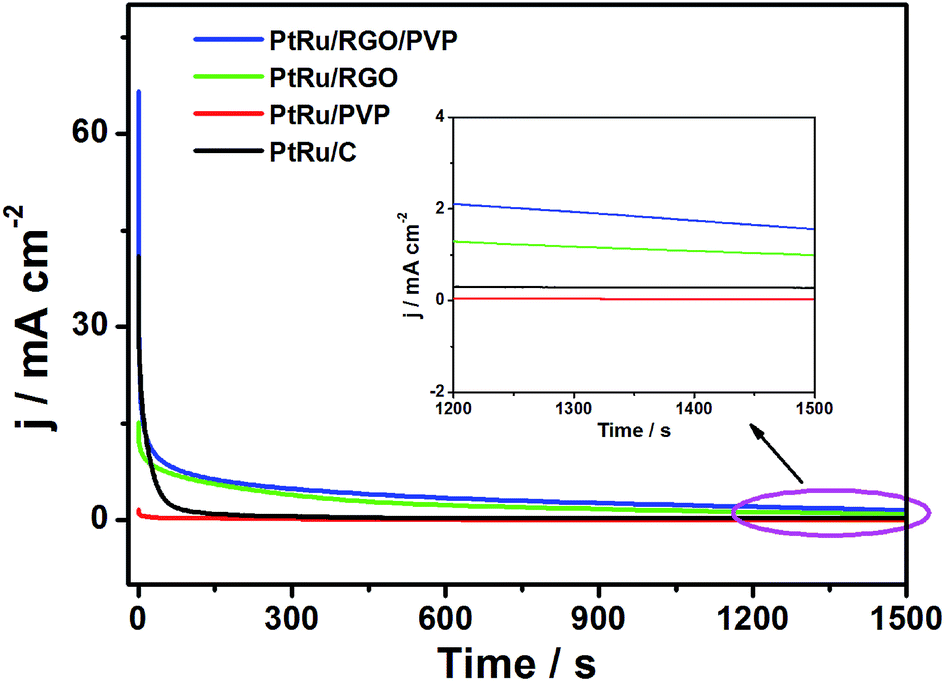 | ||
| Fig. 8 Chronoamperometric curves of PtRu/RGO/PVP, PtRu/RGO, PtRu/PVP and commercial PtRu/C catalyst in 0.5 M H2SO4 + 1.0 M CH3OH solution at 0.6 V for 1500 s. | ||
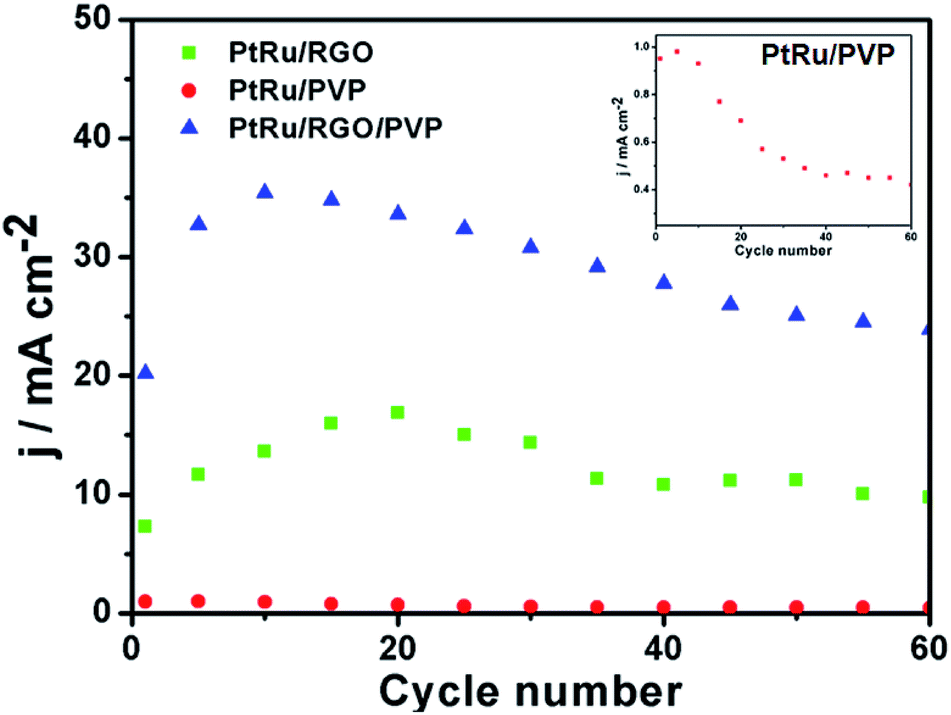 | ||
| Fig. 9 Long-term electrocatalytic stability of PtRu/RGO/PVP, PtRu/RGO and PtRu/PVP electrodes in 0.5 M H2SO4 + 1.0 M CH3OH solution at scan of 50 mV s−1. The inset shows enlarged electrocatalytic stability of spectrum for PtRu/PVP electrode. | ||
In fact, the long term of stability of a catalyst is crucial. CVs of 100 cycles for the PtRu/RGO/PVP, PtRu/RGO and PtRu/PVP electrodes were performed in 0.5 M H2SO4 + 1.0 M CH3OH solution at a scan of 50 mV s−1. The forward peak current densities as a function of cyclic scan number are shown in Fig. 9. All catalysts exhibit increased peak current densities in the initial cycle and then peak current densities slowly decrease as the number of cycles increases, which is attributed to poisoning by CO-like species and some dissolution loss of platinum.37 For PtRu/RGO/PVP catalyst, it can be seen that the forward oxidation peak current density approaches the maximum (35.4 mA cm−2) at the 10th cycle, and then decreases to 23.9 mA cm−2 after 60 cycles, with a total decrease of 32.5% relative to the maximum. However, the forward peak current densities after 60 cycles on PtRu/RGO and PtRu/PVP are reduced by 42.2% and 55.8%, respectively. These observations clearly indicate that the PtRu/RGO/PVP catalyst has much better long term stability for methanol oxidation than the PtRu/RGO and PtRu/PVP catalysts.
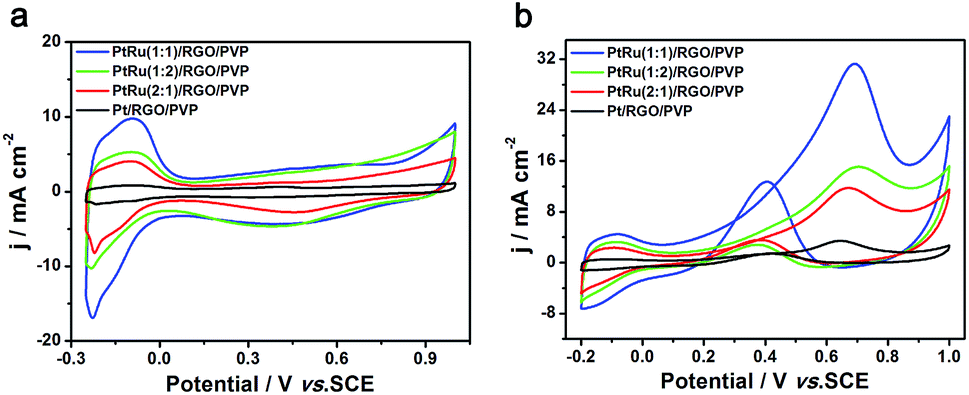 | ||
| Fig. 10 (a) CVs of Pt/RGO/PVP, PtRu(1:1)/RGO/PVP, PtRu(1:2)/RGO/PVP and PtRu(2:1)/RGO/PVP catalyst in 0.5 M H2SO4 solution at a scan of 50 mV s−1. (b) These catalysts in a mix solution of 0.5 M H2SO4 + 1.0 M CH3OH at 50 mV s−1 scan. | ||
In order to compare the electrochemical performance of different Pt/Ru atom ratios, the CVs Pt/RGO/PVP, PtRu (2:1)/RGO/PVP, PtRu (1:1)/RGO/PVP and PtRu (1:2)/RGO/PVP catalysts in 0.5 M H2SO4 solution were investigated. As shown in Fig. 10a, all catalysts had different responses to hydrogen adsorption/desorption in the −0.25 to 0.1 V region. ECSAs for PtRu (1:1)/RGO/PVP, PtRu (1:2)/RGO/PVP, PtRu (2:1)/RGO/PVP and Pt/RGO/PVP were 55.38, 29.28, 26.04 and 4.69 m2 g−1, respectively. Obviously, the PtRu (1:1)/RGO composites exhibited the largest ECSA, which is about 11.8 times greater than that of Pt/RGO/PVP. The larger electrochemical activity surface area than pure Pt can be explained by the fact that the Pt-base catalysts contained an oxophilic metal, Ru.38 The ECSA reached the highest value when the Pt NPs were combined by the Ru atom at the Pt/Au molar ratio of 1:1. Therefore, the PtRu (1:1)/RGO/PVP catalyst showed better bimetallic cooperative function and increased active sites on the electrode surface.
Fig. 10b showed the CVs for oxidation of methanol on the PtRu (1:1)/RGO/PVP, PtRu (2:1)/RGO/PVP, PtRu (1:2)/RGO/PVP and Pt/RGO/PVP modified electrodes in 0.5 M H2SO4 + 1 M CH3OH solution. An anodic current peak for the oxidation of methanol was clearly detected for the forward scan of these modified electrodes. In the reverse scan, the oxidative peak current may be ascribed to the continuous oxidation of incompletely oxidized carbonaceous species intermediates accumulated on the catalyst surface during the forward scan.39 A weak anodic peak (3.38 mA cm−2) for the Pt/RGO/PVP composites appears in Fig. 10b, which demonstrated the lowest electrocatalytic activity for methanol oxidation. The forward scan peak current densities of PtRu (1:1)/RGO/PVP (31.44 mA cm−2), PtRu (1:2)/RGO/PVP (15.19 mA cm−2) and PtRu (2:1)/RGO/PVP (11.83 mA cm−2) are, respectively, about 9.3, 2.9 and 3.5 times higher than that of Pt/RGO/PVP. We concluded that the methanol electrocatalytic-oxidation capabilities on these modified electrodes ranked as follows: PtRu (1:1)/RGO > PtRu (1:2)/RGO > PtRu (2:1)/RGO > Pt/RGO/PVP. This order is in accordance with the ECSA change trend. Our data showed that the PtRu (1:1)/RGO/PVP catalyst has the best catalytic activity among these composites.
4. Conclusions
In summary, a facile approach for synthesizing PtRu alloy catalysts by employing NaBH4 as reducing agent in the presence of PVP and RGO was investigated. With the assistance of PVP and graphene support material, the PtRu NPs were well dispersed and of smaller size; they also showed good stability and re-dispersion capability. The obtained PtRu/RGO/PVP composites exhibited a remarkably enhanced catalytic performance in the methanol oxidation reaction compared to PtRu/RGO and PtRu/PVP catalysts. This was attributed to the fact that PVP surfactant and graphene not only improved dispersion and anti-aggregation of the PtRu NPs, but also greatly enhanced electrical conductivity in the composites via synergistic transport with PtRu NPs. The PtRu/RGO/PVP catalyst with a Pt/Ru molar ratio of 1:1 exhibited the highest electrocatalytic activity. This investigation therefore demonstrated a PVP-assisted PtRu/RGO system with enhanced catalytic activity and stability, which has potential applications in DMFCs.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (51373111, 51073114 and 20933007), the Opening Project of Xinjiang Key Laboratory of Electronic Information Materials and Devices (XJYS0901-2010-01), the Project of Scientific and Technologic Infrastructure of Suzhou (SZS201207), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the Academic Award for Young Graduate Scholars of Soochow University.Notes and references
- A. S. Aricò, S. Srinivasan and V. Antonucci, Fuel Cells, 2001, 1, 133–161 CrossRef.
- H. Liu, C. Song, L. Zhang, J. Zhang, H. Wang and D. P. Wilkinson, J. Power Sources, 2006, 155, 95–110 CrossRef CAS PubMed.
- S. Basri, S. K. Kamarudin, W. R. W. Daud and Z. Yaakub, Int. J. Hydrogen Energy, 2010, 35, 7957–7970 CrossRef CAS PubMed.
- X. Zhao, M. Yin, L. Ma, L. Liang, C. Liu, J. Liao, T. Lu and W. Xing, Energy Environ. Sci., 2011, 4, 2736–2753 CAS.
- M. Cao, D. Wu and R. Cao, ChemCatChem, 2014, 6, 26–45 CrossRef CAS PubMed.
- H. Huang and X. Wang, J. Mater. Chem. A, 2014, 2, 6266–6291 CAS.
- T. J. Schmidt, H. A. Gasteiger and R. J. Behm, Electrochem. Commun., 1999, 1, 1–4 CrossRef CAS.
- L. Dong, R. R. S. Gari, Z. Li, M. M. Craig and S. Hou, Carbon, 2010, 48, 781–787 CrossRef CAS PubMed.
- H. Li, X. Zhang, H. Pang, C. Huang and J. Chen, J. Solid State Electrochem., 2010, 14, 2267–2274 CrossRef CAS.
- C. Nethravathi, E. A. Anumol, M. Rajamathi and N. Ravishankar, Nanoscale, 2013, 3, 569–571 RSC.
- H. J. Kim, S. M. Choi, M. H. Seo, S. Green, G. W. Huber and W. B. Kim, Electrochem. Commun., 2011, 13, 890–893 CrossRef CAS PubMed.
- H. P. Cong, X. C. Ren and S. H. Yu, ChemCatChem, 2012, 4, 1555–1559 CrossRef CAS PubMed.
- J. Zhao, L. Zhang, H. Xue, Z. Wang and H. Hu, RSC Adv., 2012, 2, 9651–9659 RSC.
- H. Wang, J. Du, Z. Yao, R. Yue, C. Zhai, F. Jiang, Y. Du, C. Wang and P. Yang, Colloids Surf., A, 2013, 436, 57–61 CrossRef CAS PubMed.
- S. Woo, J. Lee, S. K. Park, H. Kim, T. D. Chung and Y. Piao, J. Power Sources, 2013, 222, 261–266 CrossRef CAS PubMed.
- S. Zhao, H. Yin, L. Du, G. Yin, Z. Tang and S. Liu, J. Mater. Chem. A, 2014, 2, 3719–3724 CAS.
- X. Wang and I. M. Hsing, Electrochim. Acta, 2002, 47, 2981–2987 CrossRef CAS.
- Y. L. Hsin, K. C. Hwang and C. T. Yeh, J. Am. Chem. Soc., 2007, 129, 9999–10010 CrossRef CAS PubMed.
- E. Antolini, Appl. Catal., B, 2012, 123–124, 52–68 CrossRef CAS PubMed.
- M. Liu, R. Zhang and W. Chen, Chem. Rev., 2014, 114, 5117–5160 CrossRef CAS PubMed.
- M. Zhu, Z. Li, B. Xiao, Y. Lu, Y. Du, P. Yang and X. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 1732–1740 CAS.
- Z. Yao, M. Zhu, F. Jiang, Y. Du, C. Wang and P. Yang, J. Mater. Chem., 2012, 22, 13707–13713 RSC.
- W. Zhou, C. Zhai, Y. Du, J. Xu and P. Yang, Int. J. Hydrogen Energy, 2009, 34, 9316–9323 CrossRef CAS PubMed.
- F. Li, Y. Guo, Y. Liu, H. Qiu, X. Sun, W. Wang, Y. Liu and J. Gao, Carbon, 2013, 64, 11–19 CrossRef CAS PubMed.
- C. Zhai, M. Zhu, Y. Lu, F. Ren, C. Wang, Y. Du and P. Yang, Phys. Chem. Chem. Phys., 2014, 16, 14800–14807 RSC.
- F. Ren, H. Wang, M. Zhu, W. Lu, P. Yang and Y. Du, RSC Adv., 2014, 4, 24156–24162 RSC.
- H. L. Guo, X. F. Wang, Q. Y. Qian, F. B. Wang and X. H. Xia, ACS Nano, 2009, 9, 2653–2659 CrossRef PubMed.
- S. Guo, S. Dong and E. Wang, ACS Nano, 2010, 4, 547–555 CrossRef CAS PubMed.
- Y. Zhang, H. Shu, G. Chang, K. Ji, M. Oyamab, X. Liu and Y. He, Electrochim. Acta, 2013, 109, 570–576 CrossRef CAS PubMed.
- F. Ren, H. Wang, C. Zhai, M. Zhu, R. Yue, Y. Du, P. Yang, J. Xu and W. Lu, ACS Appl. Mater. Interfaces, 2014, 6, 3607–3614 CAS.
- J. Sun, X. Teng, J. Yang and H. Bi, J. Mater. Sci., 2013, 48, 8277–8286 CrossRef CAS.
- L. Li, Y. Wu, J. Lu, C. Nan and Y. Li, Chem. Commun., 2013, 49, 7486–7488 RSC.
- Y. Hu, H. Zhang, P. Wu, H. Zhang, B. Zhou and C. Cai, Phys. Chem. Chem. Phys., 2011, 13, 4083–4094 RSC.
- L. D. Zhu, T. S. Zhao, J. B. Xu and Z. X. Liang, J. Power Sources, 2009, 187, 80–84 CrossRef CAS PubMed.
- A. Barinov, O. B. Malcioglu, S. Fabris, T. Sun, L. Gregoratti, M. Dalmiglio and M. Kiskinova, J. Phys. Chem. C, 2009, 113, 9009–9013 CAS.
- J. Zhao, L. Zhan, H. Xue, Z. Wang and H. Hu, RSC Adv., 2012, 2, 9651–9659 RSC.
- S. J. Yoo, T. Y. Jeon, K. S. Kim, T. H. Lim and Y. E. Sung, Phys. Chem. Chem. Phys., 2010, 12, 15240–15246 RSC.
- A. V. Palenzuela, E. Brillas, C. Arias, F. Centellas, J. A. Garrido, R. M. Rodríguez and P. L. Cabot, J. Power Sources, 2013, 225, 163–171 CrossRef PubMed.
- C. Pan, L. Qiu, Y. Peng and F. Yan, J. Mater. Chem., 2012, 22, 13578–13584 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Particle-size distribution histograms and enlarged XRD pattern of samples, EDX spectrum of PtRu/RGO/PVP. See DOI: 10.1039/c4ra07742c |
| This journal is © The Royal Society of Chemistry 2014 |