
Macroporous and nanometre scale fibrous PLA and PLA–HA composite scaffolds fabricated by a bio safe strategy
Aurelio Salerno*a,
Mar Fernández-Gutiérrezbc,
Julio San Román del Barriobc and
Concepción Domingo Pascuala
aInstitute of Materials Science of Barcelona (ICMAB-CSIC), Campus de la UAB s/n, Bellaterra, 08193, Spain. E-mail: asalerno@icmab.es
bInstitute of Polymer Science and Technology, CSIC, Madrid 28006, Spain
cCIBER-BBN, Health Institute Carlos III, Spain
First published on 11th November 2014
Abstract
The present study deals with the design, fabrication and characterization of porous scaffolds for tissue engineering made of polylactic acid (PLA) and PLA containing hydroxyapatite (HA) nanoparticles. The main novelty relies on the fact that the fabrication of the scaffolds has been achieved avoiding totally the use of toxic chemicals. In particular, the scaffolds are obtained by combining both thermal induced phase separation (TIPS) using ethyl lactate (EL) solutions and supercritical CO2 (scCO2) drying processes. Furthermore, gelatin particles have been used as a leachable porogen and combined with the previous processes to improve the control of the pore structure features of the scaffolds. The results show that the developed technique allows for the fabrication of porous PLA scaffolds with HA concentrations up to 30 wt%. Furthermore, these scaffolds are characterized by an overall porosity as high as 98% and a double scale pore structure. In particular, the appropriate control of the TIPS process and the scCO2 drying allowed for the development of nanoscale fibrous PLA and PLA–HA structures starting from PLA/EL solutions. Concomitantly, the proper selection of the size range of gelatin particles as well as their spatial distribution in the mixture allowed imprinting an interconnected network of large pores inside the scaffolds.
Introduction
The search for novel materials and manufacturing techniques for scaffold design and fabrication is still a major challenge in tissue engineering (TE). In the TE approach, the scaffold serves as a provisional three-dimensional (3D) template promoting cell growth and new tissue development.1,2 To achieve these aims, ideal TE scaffolds must be biocompatible and biodegradable with a degradation rate matching the rate of new tissue formation in vitro and/or in vivo.3 Furthermore, the scaffolds must have a network of interconnected pores enabling 3D adhesion, proliferation and colonization of cells.4Biodegradable polymers, mainly polyesters such as poly(lactic acid) (PLA) and polycaprolactone (PCL), have been widely used to develop 3D porous scaffolds from both solution- and melt-based processes.5–7 Indeed, scaffolds prepared starting from these synthetic materials demonstrated good biocompatibility and degrade into non-toxic components with a controllable degradation rate.4 Research on biomaterials for scaffolds fabrication has also been focused on bioactive natural and synthetic ceramics.4,7–9 These materials, such as hydroxyapatite (HA), β-tricalcium phosphate and bioactive glasses, have been mainly developed for bone TE scaffolds due to their chemical similarity to the inorganic phase of bone.4,8 The bioactive nature of these materials is ascribable to their biodegradation and consequent ions release.9 For instance, it was observed that ions formed in the dissolution products of bioactive glasses can stimulate the expression of several genes of osteoblasts.9 The chemical composition and both the micro and nanostructure of these materials can be engineered to tailor the release of suitable ions for enhancing scaffold vascularisation,9 as well as for the repair/regeneration of soft tissues such as nerve.10
Development of porous composite scaffolds is attractive as it takes the advantages of two or more types of materials, which can be combined to suit better the chemical and physical demands of the host tissue.11 By taking advantage of the formability of polymers and by including a controlled-amount of a bioactive ceramic phase, porous scaffolds with enhanced morphology and proper structural properties can be achieved.12,13 Concomitantly, polymer–ceramic composites can overcome the poor bioactivity of most polymers by providing a biomimetic environment for cells and tissues.14
A critical step when designing 3D porous composite scaffolds for TE is related to the appropriate selection of the processing technique. Ideally, scaffolds manufacturing should allow for the control and design of micro or nanometer scale structures within polymeric composites. These include the control of the overall porosity of the scaffolds, as well as its pore size distribution, interconnectivity and texture.2,3,12 Furthermore, processing techniques able to induce the formation of a nano-scale fibrous architecture on the pore walls of the scaffold are strongly required. Indeed, owing to their high surface-to-volume ratio and morphological and mechanical features, the nanofibrous structure is expected to enhance cell adhesion, proliferation and differentiation.12,15 For instance, nanoscale fibrous scaffolds have been appropriately fabricated for bone,15 nerve,16 blood vessel17 and other soft tissue18 regeneration. Achieving these goals by means of a bio safe process is also mandatory for the final clinical implementation of porous composite scaffolds.
We have recently reported a novel biosafe process based on a thermally induced phase separation (TIPS) technique for the design and fabrication of nanometrescale fibrous polylactic acid (PLA) aerogel-like materials with controlled morphology and structural properties.19 The process involved the use of ethyl lactate (EL) to process PLA and a supercritical CO2 (scCO2) solvent extraction technique for gel drying. EL is a USA Food and Drug Administration approved additive in the food industry, as it does not has potential health risks.20 Furthermore, this solvent is biodegradable and can be produced from natural resources. Concomitantly, the scCO2 drying process allows obtaining polymeric aerogels from PLA/EL solutions avoiding the collapse of the fibres network during solvent extraction.19
In this work, we further improve the fabrication route previously developed19 by investigating the fabrication of PLA and PLA–HA nano-composite scaffolds. Herein, we report an easy and tuneable approach for the control of the pore structure features of the scaffolds by combining TIPS and porogen leaching techniques. The developed process aims to obtain PLA and PLA–HA nanocomposite scaffolds with an interconnected network of macropores and a nanoscale fibrous pore wall morphology. Gelatin particles of two different size distributions, namely 200–400 and 400–600 μm, were used as the porogen agent.21 Gelatin particles combination with the polymeric solution followed three different strategies. The strategy 1 involved the simple dispersion of the gelatin particles inside the polymeric solution followed by TIPS and scCO2 drying processes. The strategies 2 and 3 involved the preparation of a pre-sintered gelatin template, followed by its infiltration with the polymeric solution by either centrifugation (strategy 2) or low vacuum impregnation (strategy 3) approach. The as obtained scaffolds were characterized to assess their chemical composition and thermal properties, as well as their morphology, pore structure features, mechanical properties and in vitro biocompatibility.
Experimental section
Materials
The used PLA has an 80/20 L/dL ratio, a molecular weight of 200 kDa and an inherent viscosity at midpoint of 3.8 dL g−1 and was provided by Purac Biochem (Code PLDL8038, Gorinchem, The Netherlands). HA nanopowder (Code 677418, Sigma-Aldrich, Madrid, Spain), with a mean particle size lower than 200 nm and a surface area of 9.4 m2 g−1, was selected for the preparation of the PLA–HA composites. EL (Code 77367, photoresist grade; purity ≥99.0%) and ethanol (Code 02860, 99.8%) were provided by Sigma-Aldrich (Madrid, Spain) and used without further purification. Gelatin particles (Code 4078, Merck, Darmstadt, Germany) were used as the particulate porogen. CO2 (99.95 wt%, Carburos Metálicos) was used as the drying agent.Measurement of the gelation point curves
In this work, liquid–liquid TIPS process was used to prepare the nanoscale fibrous PLA and PLA–HA composite scaffolds. This process involves first the cooling of the polymeric solution down to the gelation point. By lowering the temperature, the polymer–solvent affinity decreases and the system separated into a polymer-rich and a polymer-lean phases. The interpenetration of these two phases is responsible for the gelification of the solution. Measurements were carried out to assess the gelation point of prepared PLA/EL and PLA–HA/EL solutions. The PLA/EL solution was prepared by dissolving the polymer in EL at a concentration of 5% w/v and 70 °C under magnetic stirring for 8 h. For composites preparation, the HA nanoparticles were first dispersed in EL to form a homogeneous suspension. Particles dispersion was achieved by magnetic stirring (30 min) followed of sonication (30 min). These steps were repeated twice to ensure the achievement of a homogeneous solution by reducing the amount of aggregated particles. Subsequently, the polymer was added to the solution and the system was maintained under magnetic stirring at 70 °C for 8 h. Different HA particles concentrations ranging from 10 to 30 wt% with respect to the amount of polymer were used based on literature investigations of convenient percentages for similar systems.22 For the gelation point measurement, 1 mL of each solution was sealed in a 2 mL glass tube and placed in a water bath pre-heated at 50 °C. The temperature was then slowly cooled in steps of 0.5 °C giving an equilibrium time of 15 min. At each step, the vial was inverted vertically and the gelation point defined as the temperature at which the solution ceased to flow.Fabrication of PLA and PLA–HA composite scaffolds by TIPS combined to scCO2 drying
Nanometre scale fibrous scaffolds were fabricated by the TIPS method combined with a scCO2 drying process and starting from the PLA and PLA–HA solutions previously described. To this purpose, 0.5 mL of solution were added to cylindrical aluminium moulds, measuring 6 mm in diameter and 20 mm in height, followed by cooling to 6 °C to induce phase separation and solution gelation. After 3 h of gelation, the samples were removed from the mould and soaked in excess of cold ethanol for solvent exchanging. This step induced the polymer precipitation and nanometre scale fibres formation. The solvent was subsequently interchanged with fresh ethanol for three times to ensure the complete removing of the EL. After that, the as obtained PLA and PLA–HA alcogels were dried using scCO2. Drying was carried out in a high-pressure autoclave (TharDesign, Pittsburgh, USA) with a cylindrical section and a total volume of 114 mL. The samples were placed inside of the autoclave on the top of a metallic support to allow for the addition of a magnetic stirrer at the bottom of the vessel. This configuration is expected to improve fluids mixing and to reduce the equilibrium time. The temperature was then raised up to 39 °C and liquid CO2 was pumped inside the vessel up to a pressure of 19 MPa, thus ensuring the achievement of supercritical conditions without crossing the glass transition temperature of PLA. Samples were held at these conditions for 90 min. Pure CO2 was finally flushed inside the vessel for 15 min and the vessel was depressurized slowly to the ambient pressure.Fabrication of PLA and PLA–HA composite scaffolds with a pre-designed network of large-pores by combining TIPS, gelatin particles leaching and scCO2 drying
The preparation of porous scaffolds characterized by a network of large pores and a nanometre scale fibrous architecture was obtained by using gelatin particles as the porogen agent in combination with TIPS and scCO2 drying. The gelatin particles have been used as provided by the manufacturer. However, to control the size of the large pores in the scaffolds, gelatin particles were sieved into two different size ranges, namely 200–400 μm and 400–600 μm. As reported in the scheme of Fig. 1, three different strategies were used, each one differing basically on the assembly of the gelatin particles and their mixing with the polymeric solution. In the strategy 1, the sieved particles were manually mixed with the polymeric solution (0.8% w/v). Next, the as prepared mixture was placed in an aluminium mould, measuring 6 mm in diameter and 15 mm in height, and slightly compressed to promote the compaction of the mixture and the contact between adjacent gelatin particles. Solution gelation was further induced by keeping the samples at 6 °C for 3 h. Once the setting of the gel was achieved (Fig. 1), the EL and gelatin particles were sequentially extracted by soaking the gel first in ethanol for 1 h at room temperature and then in water at 40 °C for 2 days. Finally, the scaffolds were processed by means of a scCO2 drying process as previously described. To improve the control in the large pores interconnection, in the strategies 2 and 3 the gelatin particles were pre-sintered into a three dimensional template structure and, then, the polymeric solution was infiltrated into the void space of the template. Gelatin particles sintering was achieved by first soaking a mould containing the compacted particles into a 90/10 ethanol–water solution for 5 min, followed by the extraction of the excess of solution and the drying of the sample at 100 °C for 10 min (SEM images of Fig. 1). In the strategy 2, the infiltration of the gelatin template was achieved by dipping the polymeric solution onto the template, followed by centrifugation at 100 rpm for 2 min at room temperature. Conversely, in strategy 3, the template was soaked in excess of the polymeric solution at 60 °C and the system kept at 0.01 MPa for 5 min (Fig. 1). The as prepared samples were then processed by TIPS and scCO2 drying processes, as previously described.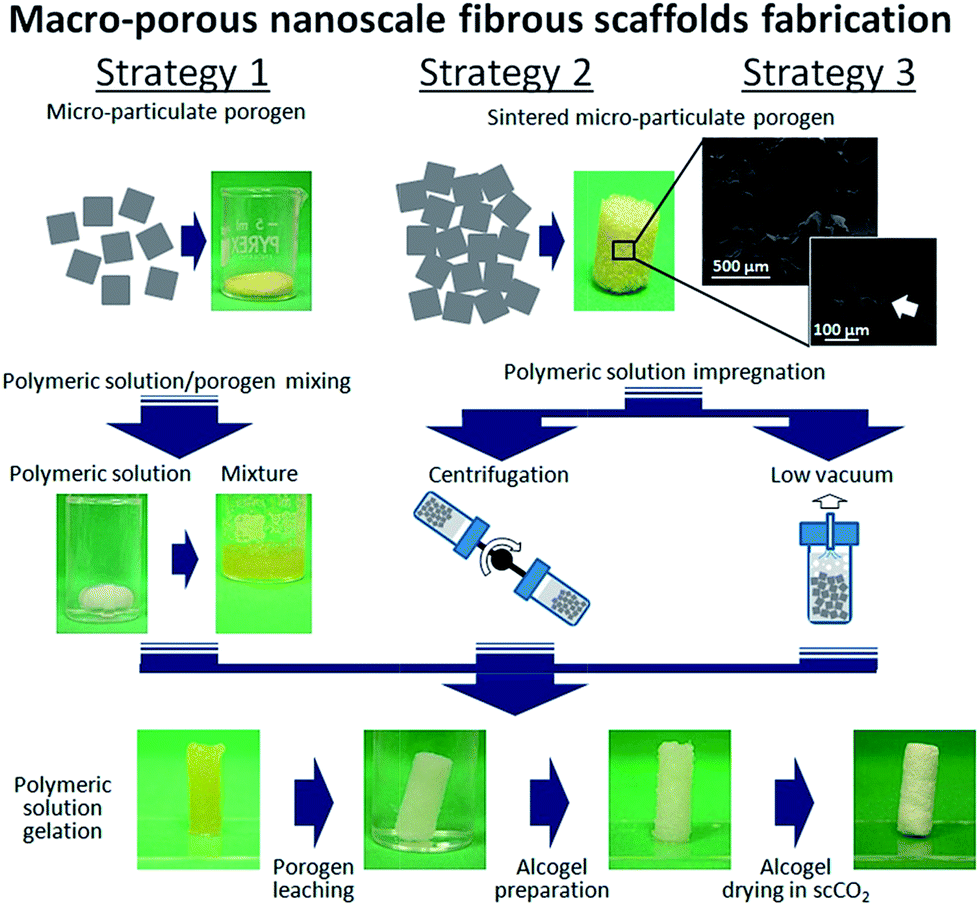 | ||
| Fig. 1 Scheme of the different strategies used for the preparation of porous PLA and PLA–HA composite scaffolds by means of the TIPS, gelatin leaching and scCO2 combined processes. | ||
Scaffolds characterization
Thermogravimetric (TGA-DTGA) and differential scanning calorimetric (DSC) analyses were carried out on PLA and PLA–HA composite scaffolds to assess the effect of the TIPS process and materials composition on the HA loading efficiency, as well as on the thermal stability and crystalline properties of the samples. TGA-DTGA experiments were carried out with a TGA STA 449 F1 Jupiter (NETZSCH, Selb, Germany) in the 30 to 600 °C temperature range by increasing the temperature at 10 °C min−1 under an inert atmosphere. The degradation temperature (Tdeg) was assessed as the maximum of the DTGA curve in the 300–400 °C temperature range. The HA loading in the composite scaffold was determined by measuring the residue weight at 600 °C and subtracting the as obtained value by the residue weight of the neat PLA scaffold. The scaffolds were further tested on a DSC8500 apparatus (Perkin Elmer, Massachusetts, USA) equipped with a liquid N2 controller CRYOFILL. Samples were first equilibrated at 0 °C for 5 min and then heated up to 150 °C at a scanning rate of 10 °C min−1 under an inert atmosphere. The glass transition (Tg) and melting peak (Tp) temperatures and the crystalline fraction (χC) of the scaffolds were determined from the DSC tests.Scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDX) were carried out to assess the morphology of the scaffolds and their elemental composition. Samples were cross-sectioned and gold sputtered prior to analysis with a SEM-EDX equipment (QUANTA 200F FEG-ESEM, FEI, The Netherlands).
Transmission electron microscopy (TEM) analysis was used to evaluate the morphology and distribution of the HA filler within the scaffolds. Samples were first embedded in a 10% w/v gelatin solution at 40 °C and maintained under vacuum for 30 min, and then frozen at −15 °C overnight. The samples were subsequently sectioned with a Leica FC6 cryostat (Wetzlar, Germany) at −80 °C to obtain 100 nm slices. The slices were analyzed at an acceleration voltage of 100 kV with a JEM-2011 (Tokyo, Japan).
The porosity of the scaffolds was assessed by two different gravimetric measurements. In the first method, the overall porosity was determined from the mass and volume measurements.23 The mass of the scaffolds was measured by using a high precision balance (10−4 g, CPA225D, Sartorius, Gottingen, Germany), while the volume was determined by a geometrical calculation using a calliper. The as obtained values were then used to determine the density of the scaffolds. These values were corrected by considering the effective HA loading, as measured by the TGA tests. In the second method, the porosity of the scaffolds was determined by using the following eqn (1):
| porosity% = (mW − mD)/[(mD/ρB) + mW] × 100 | (1) |
The biocompatibility of the scaffolds was assessed in vitro by using human osteoblast cells (HOb 406-05f, ECACC, Sigma Aldrich, Madrid, Spain). Scaffolds for cell culture experiments were disinfected by soaking them in absolute ethanol and further washed with sterile PBS (phosphate buffer saline, pH 7.4, Sigma) and culture medium during 24 h. The culture medium was Dulbecco's modified Eagle's medium enriched with 4500 mg mL−1 of glucose (DMEM, Sigma) supplemented with 10% foetal bovine serum (Gibco), 200 mM L-glutamine, 100 units per mL penicillin, 100 mg mL−1 streptomycin and 1% non-essentials amino acids (NEEA) modified with HEPES (all from Sigma Spain).
Cells were statically seeded onto the scaffolds at a density of 1 × 105 cell per mL. The cell-scaffold constructs were subsequently placed into 96-well culture plate and maintained in excess of culture medium up to 7 days, and the culture medium changed at selected time intervals. Fibrin gel was used as the control sample for cell culture tests. Quantitative analysis of cell adhesion and proliferation onto the scaffolds was carried out by means of the Alamar Blue test. At pre-determined culture times, 100 μL of Alamar Blue dye (Serotec, 10% Alamar Blue solution in phenol red free DMEM medium) was added to the cell-scaffold constructs and the samples were incubated for 4 h. Subsequently, 100 μL of the reaction media for each test sample was extracted and transferred to a 96-well plate. The fluorescence emission was measured at 590 nm ex and 630 nm em on a Biotek Synergy HT. The remaining samples were washed with PBS twice to remove any residual reagent, and 1 mL of the culture medium was added to continue cell culture experiments. This step was done at 1, 3 and 7 days. Analysis of variance (ANOVA) of the results was performed with respect to control with a p < 0.05 significance level.
Results and discussion
Gelation point of PLA/EL and PLA–HA/EL solutions
The fabrication of porous PLA and PLA–HA composite scaffolds by TIPS technique requires the cooling of the homogeneous solution down to the gelation point. Here, the optimization of the conditions for samples gelation were achieved based on our previously reported work on PLA/EL solution19 and by the determination of the gelation point of the solution as a function of the HA loading. The results of these tests (not shown) evidenced that the addition of the inorganic filler increased the gelation point of the polymeric solution. In particular, we observed that the gelation temperature increased from 30.5 °C in the case of neat PLA up to 32 °C for the sample with 30 wt% of HA nanoparticles. This effect can be ascribed to the fact that the inorganic phase acted as a nucleating agent for the formation of stable polymeric nuclei, thus, accelerating their crystallization and the final setting of the gel.24Properties of PLA and PLA–HA scaffolds prepared via TIPS and scCO2 drying processes
The preparation of porous nanometre scale fibrous PLA and PLA–HA composite scaffolds was achieved by using TIPS combined with scCO2 drying process. The low and high magnification SEM micrographs of the PLA scaffolds reported in Fig. 2A and C, respectively, confirmed that the samples are characterized by a nanometre-scale fibrous structure. As shown in Fig. 2B the addition of the HA nanoparticles did not affect the formation of the nanometre-scale fibres, while the inorganic filler was quite homogeneously distributed onto the surface of the polymeric fibres (white arrows in Fig. 2D). These results clearly demonstrated that the solution-based processing route developed for the preparation of PLA–HA composite scaffolds is a good choice to achieve a homogeneous distribution of the inorganic filler into the polymeric matrix. Fig. 2E and F reported the elemental composition assessed by EDX analysis and the TEM images of the PLA and PLA–HA composite containing 30 wt% of nanoparticles. The results of the EDX analysis indicated that the HA was characterized by a Ca/P ratio of 1.78, close to the stoichiometric HA composition.23 Furthermore, in agreement to SEM observations, the TEM micrographs reported in Fig. 2F showed that the HA nanoparticles possess a round morphology and the majority of these particles are characterized by a size below 100 nm. TEM analysis also evidenced the high particles dispersion inside of the polymeric fibres and suggested their exposing on the fibres surface (Fig. 2F). | ||
| Fig. 2 Characterization of PLA and PLA–HA composite scaffold with 30 wt% of nanoparticles: SEM micrographs of (A and C) PLA and (B and D) PLA–HA; (E) EDX analysis showing the elemental composition of PLA and PLA–HA; and (F) TEM images of PLA–HA. | ||
In Fig. 3 and Table 1 are reported the results of the thermal characterization of the PLA and PLA–HA nanometre scale fibrous scaffolds. The TGA-DTGA analysis provided important information about the thermal stability of the samples as well as the retained amount of HA nanoparticles inside the scaffolds. The addition of HA enhanced the thermal stability of the polymeric matrix, as evidenced by the increase of the Tdeg from 348.5 °C for neat PLA up to 363.1 °C for the PLA–30HA sample (Fig. 3A). This result is in agreement with those reported in the literature for other composite systems and it is ascribable to the superior insulation and mass-transport barrier properties of the inorganic nanoparticles that slow down the decomposition of the polymeric matrix.25 The retained HA nanoparticles inside the PLA matrix was higher than 85% for the 10 and 20 wt% HA loaded samples and increased up to more than 92% for the 30 wt% HA loaded sample.
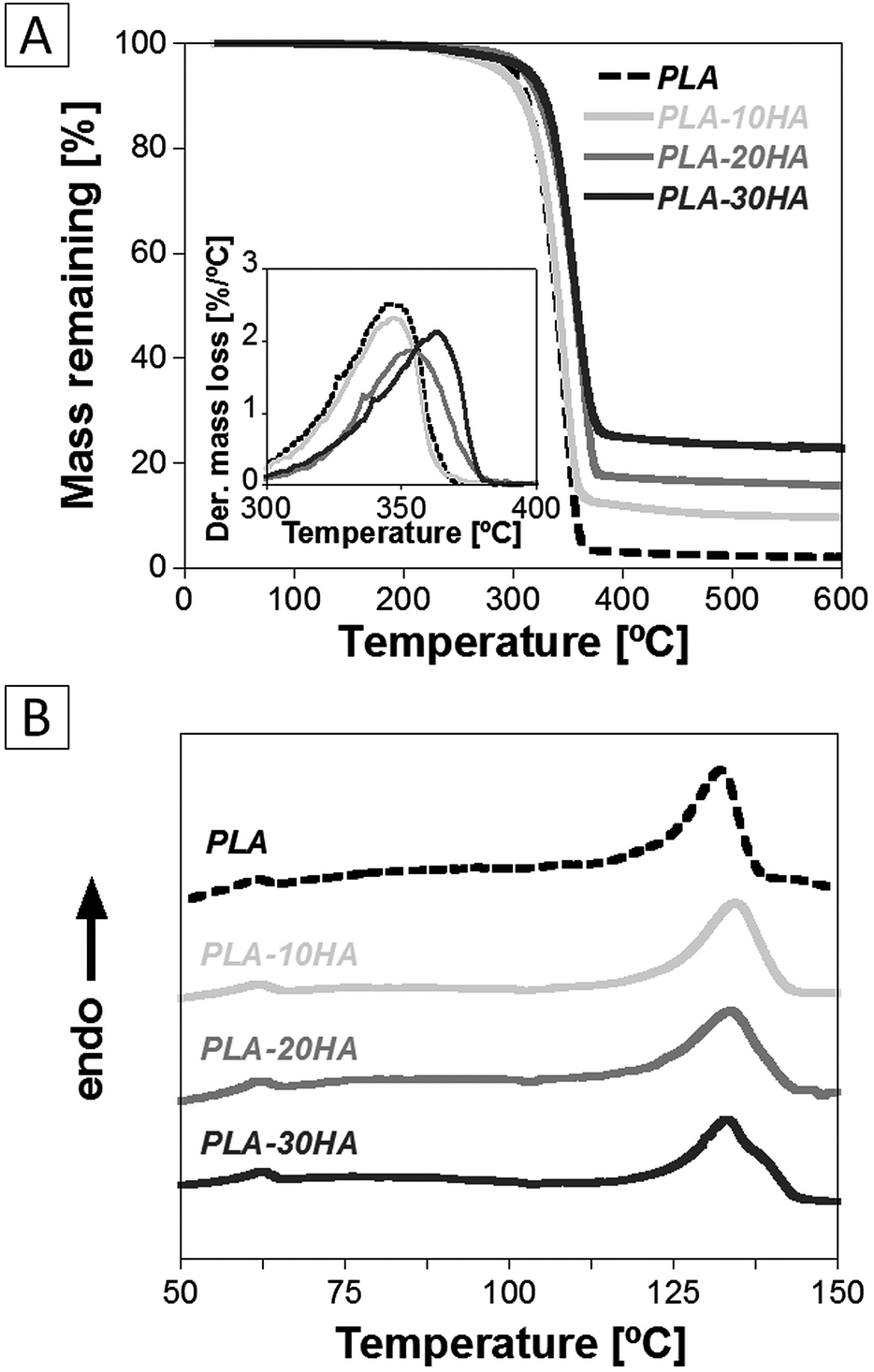 | ||
| Fig. 3 Results of (A) TGA-DTGA and (B) DSC analysis of the nanometre-scale fibrous PLA and PLA–HA composite scaffolds. | ||
| Sample | Tga [°C] | Tpb [°C] | χCc [%] | Tdegd [°C] | Res600 °C [%] | HA [%] |
|---|---|---|---|---|---|---|
| a Glass transition temperature.b Melting peak temperature.c Crystalline fraction.d Degradation temperature. | ||||||
| PLA | 59.8 | 129.6 | 20.5 | 348.5 | 2.0 | — |
| PLA–10HA | 60.6 | 131.9 | 21.9 | 348.5 | 9.6 | 85.8 |
| PLA–20HA | 60.3 | 131.2 | 21.5 | 351.9 | 15.8 | 85.2 |
| PLA–30HA | 60.7 | 131.0 | 22.5 | 363.1 | 22.8 | 92.7 |
In Fig. 3B the DSC curves of the first heating scan of the PLA and PLA–HA composite scaffolds are reported. The obtained data from the melting behaviour are listed in Table 1. All samples were characterized by Tg values close to 60 °C, while Tp and χC values slightly increased after the incorporation of HA within the polymer. The semi crystalline nature of the scaffolds prepared confirmed that PLA crystallization is a key aspect for the successful development of a nanometre scale fibrous structure during the TIPS process. Indeed, it was reported that in the early stage of the PLA nanometre scale fibres formation induced by the TIPS process, the polymer condensed first in the form of amorphous nanoparticles of about 20 nm in diameter and then it crystallized from the gel. This second step occurred trough the formation of a nanometre scale fibrous structure, which is initiated from the previously nucleated particles and radial grew outward.26 The morphological characterization evidenced that the gelation temperature used, more than 20 °C lower than the gelation point of the solution, was sufficient to ensure that crystallization occurred after phase separation and, therefore, to achieve the nanometre scale fibres. Furthermore, the addition of the HA nanoparticles in the polymeric solution slightly lowered the gelation point and consequently increased the crystallinity of the samples (Table 1), while this effect did not affect the formation of the fibrous architecture.
Another critical aspect regarding the fabrication of nano scale fibrous polymeric scaffolds is related to the process of gel drying. Indeed, owing to the high specific surface area and the tiny fibrous morphology, drying the gel in air often provides a white and dense collapsed xerogel. This effect depends on the capillary forces exerted on the pore walls by the evaporating liquid. ScCO2 drying overcomes this drawback drying the gels through the formation of a supercritical solution with the liquid solvent inside the pores, typically ethanol.19 The scCO2–solvent mixture shows no surface tension and can be eliminated from the sample by venting the pressure vessel to the ambient pressure. Moreover, the selection of the appropriate temperature and pressure conditions is important to avoid crossing the glass transition temperature of the polymer and passing through the liquid state of the supercritical mixture during venting.
Properties of macroporous nanometre scale fibrous PLA and PLA–HA composite scaffolds
In designing porous scaffolds for tissue engineering, one of the most important aspects is the achievement of a porous structure with pores both large enough and sufficiently interconnected to promote cell colonization and infiltration in 3D.1–4 To induce the formation of controlled networks of large pores inside the PLA and PLA–HA composite scaffolds previously prepared, the TIPS and scCO2 drying combined process was improved by using gelatin particles of controlled size distribution as additional porogen. The selection of gelatin takes advantages on the evidences that this polymer is almost insoluble in EL and can be mixed with the polymeric solution before gelation. Furthermore, gelatin is a well known biocompatible polymer and, consequently its use as a porogen agent is in line with the bio safe nature of the scCO2 process developed in this work. The control of the size of the pores was achieved by selecting gelatin particles with two different size distributions: 200–400 and 400–600 μm.In Fig. 4, it is reported the morphology of porous PLA scaffolds prepared following the three different strategies highlighted in Fig. 1. As shown, the scaffolds are provided of pre-designed networks of large pores and nanometre scale fibrous pore wall textures. Furthermore, the morphology of the scaffolds can be adequately adjusted depending on the selected strategy and the size range of gelatin particles used. In particular, for all of the systems we observed the increase of the size of the large pores with the increase of the size range of gelatin particles from 200–440 to 400–600 μm. SEM observation of the scaffolds shown in Fig. 4 also suggests an increase of the percentage of large pores on the walls and a decrease of large-pore interconnections with the increase of the size of the starting porogen. These effects are ascribable to the decrease of the contact points between the gelatin particles as a consequence of the increase of their size. Most importantly, close observation of the pore wall architecture of the scaffolds revealed the presence of a nanometre scale fibrous structure close to the structure reported in Fig. 2C. If compared to the PLA scaffolds obtained following strategy 1, those prepared by using strategies 2 and 3 evidenced thinner pore walls and enhanced interconnectivity among the large pores.
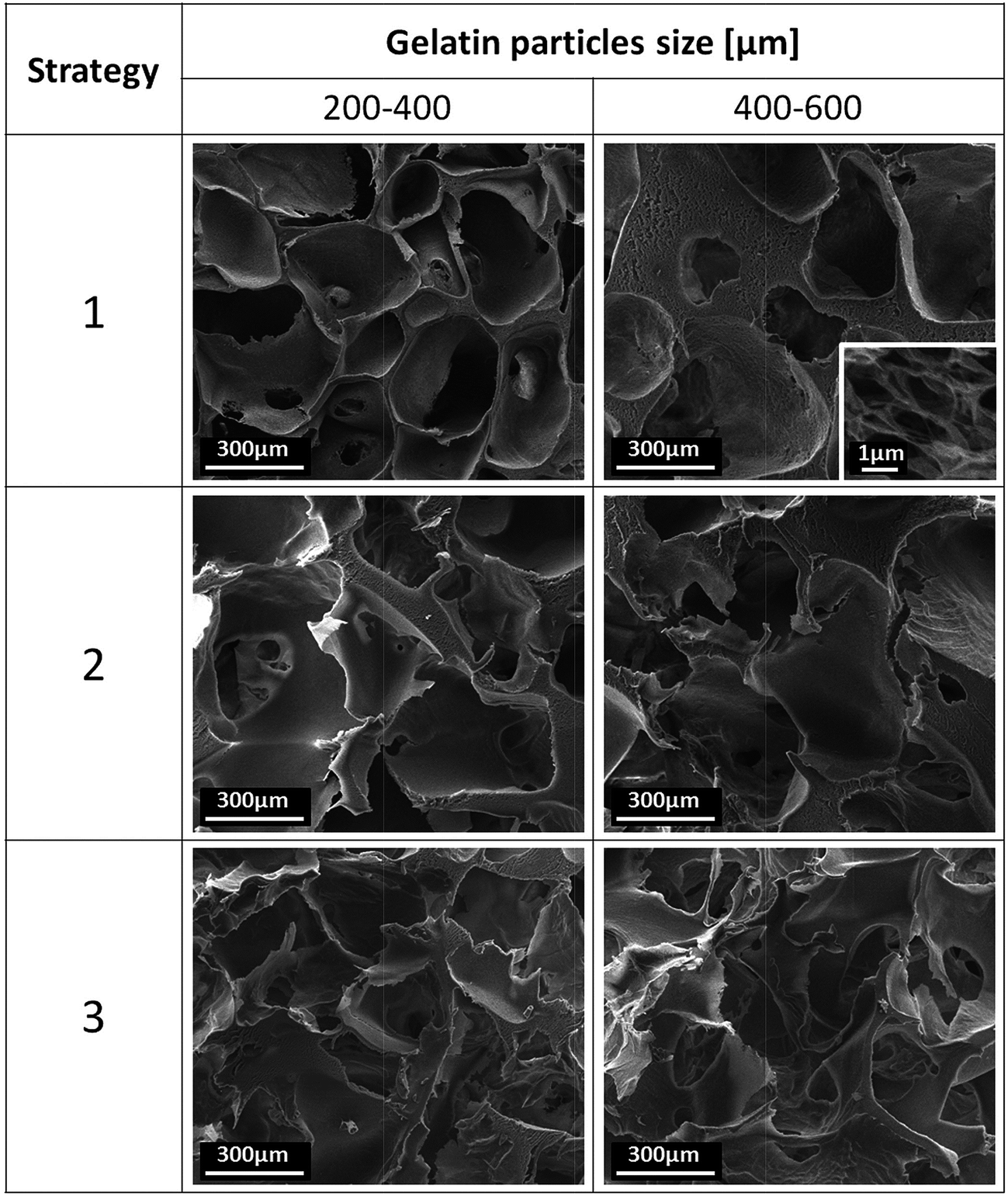 | ||
| Fig. 4 SEM images showing the morphology of PLA scaffolds prepared by means of the three different strategies used in this work. | ||
The porosity and mean size of the large-pore voids of PLA scaffolds as a function of the fabrication route are reported in Fig. 5. As shown, all of the scaffolds evidenced porosity values higher than 95%, while the scaffolds prepared following strategies 2 and 3 were provided of the highest porosity values, up to 97%. As evidenced in Fig. 5B, the mean large-pore size of PLA scaffolds prepared following strategy 1 increased from 273 ± 39 to 439 ± 80 μm when the size range of gelatin particles increased from 200–440 to 400–600 μm. This increase was less marked in the case of scaffolds prepared by using the pre-sintered gelatin template as porogen, since for these samples the mean large-pore size was equal to 350 μm when 200–400 μm gelatin particles were used and equal to 420 μm for gelatin particles in the 400–600 μm size range. This effect can probably be ascribed to the fact that, in such case, the sintering step can induce the complete fusion of adjacent gelatin particles and promote the formation of larger pores, especially when small gelatin particles were used. For all of the studied samples, the large pore walls were provided of a nanometre scale fibrous architecture with fibres size in the 100–200 nm range.
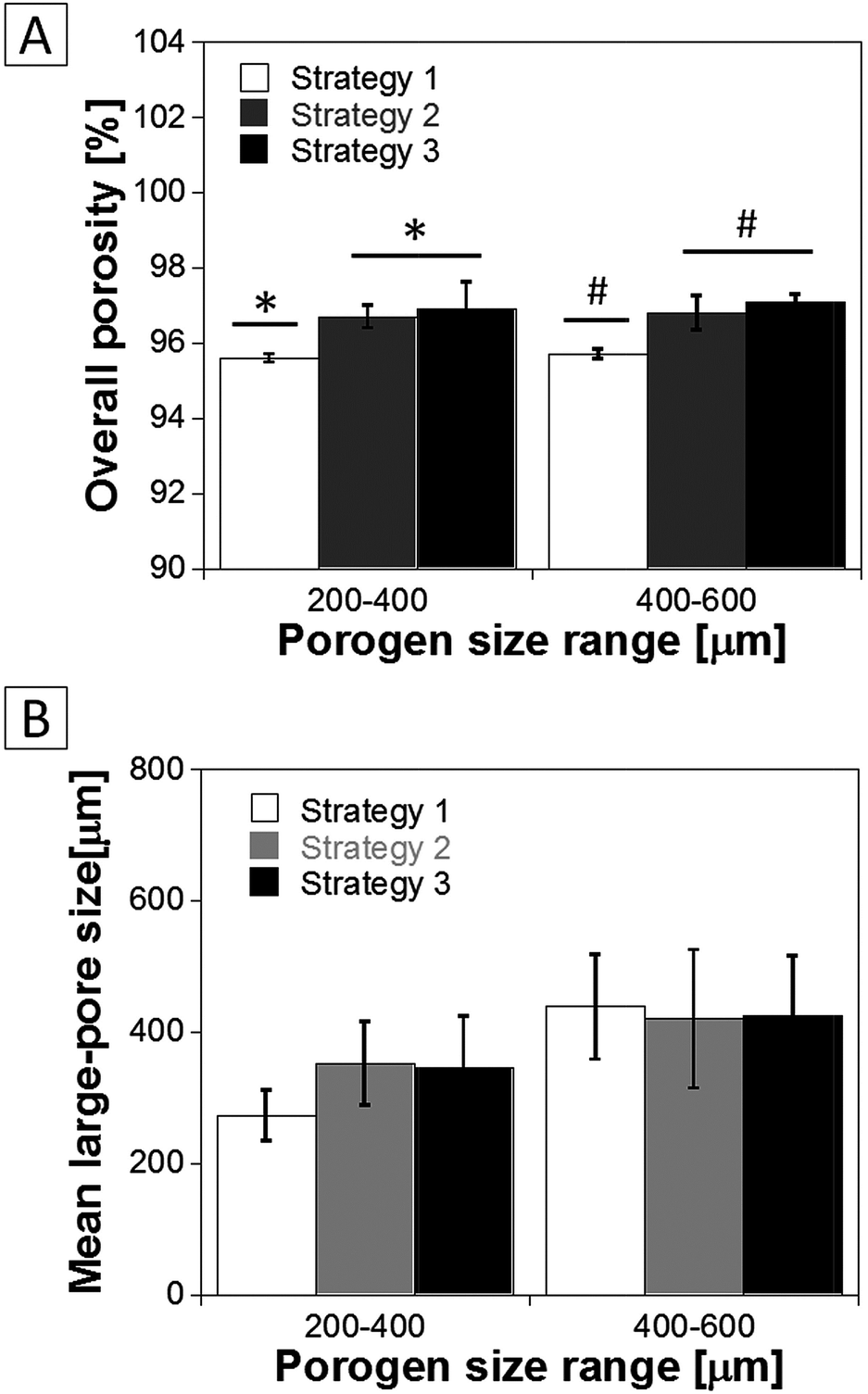 | ||
| Fig. 5 Porosity and mean size of large pores of PLA scaffolds prepared by means of the three different strategies used in this work. | ||
These considerations were supported by the results of confocal analysis reported in Fig. 6, where the 3D reconstruction of PLA scaffolds prepared following strategies 1 and 3 are compared. As shown, the size of the pores increased with the increase of the size of gelatin particles used. Samples prepared following strategy 3 were characterized by less definite pore walls. Confocal analysis allows also quantifying the roughness and the mean area of the pores of the scaffolds. We observed that the roughness increased with the increase of the size of the pores, while the highest value was observed for the scaffold prepared following strategy 3. For instance, for the PLA scaffold prepared by using 200–400 μm gelatin particles, the roughness increased from 70 to 90 μm for switching from strategy 1 to 3. The measured values of large pore surface area of scaffolds prepared using 200–400 μm were 64 × 103 μm2 for scaffolds prepared following strategy 1 and 87 × 103 μm2 for scaffolds prepared following strategy 3. These values corresponded to spherical pores of mean size equal to 285 and 333 μm, respectively, and therefore are in good agreement with previously reported data obtained by means of Image J analysis.
 | ||
| Fig. 6 Results of confocal characterization of PLA scaffolds prepared by using gelatin particles with (A and C) 200–400 μm size and (B and D) 400–600 μm size. Images (A and B) refer to strategy 1 while images (C and D) refer to strategy 3. | ||
There are several reported studies about the use of particles-fused templates for the control of the interconnectivity of porous scaffolds prepared by a porogen leaching technique.27–29 The main advantage of using this approach for scaffolds fabrication is the possibility to control finely the interconnection of the pores by directing particles fusion. For instance, it has been demonstrated that the shape of NaCl porogen particles can be shifted from cubic to spherical by simply increasing the sintering time, which, consequently, enhanced porous scaffolds interconnection were achieved because of the increased contact area among adjacent particles.27 Similar results were achieved by increasing the sintering time of a paraffin spheres template or by increasing the pressure applied during elvacite beads compaction.28,29
The morphological characterization of our scaffolds supported these finding and also demonstrated that this process can be efficiently combined with TIPS and scCO2 drying for preparing porous PLA scaffolds. Indeed, by using a pre-sintered gelatin mould, instead of mixing the particulate porogen with the polymeric solution, porous PLA scaffolds with enhanced pore interconnectivity were achieved. The enhanced compaction of the porogen particles after sintering reduced the void space available for the infiltration of the polymeric solution and consequently induced a slight increase of the overall porosity of the scaffolds.
In order to optimize the scaffold fabrication, we also compared the effect of two different strategies for the infiltration of the polymeric solution into the gelatin template on the final scaffold properties. As a result, we observed that the careful control of the operating conditions during the process allowed for the almost complete penetration of the polymeric solution in the void space of the pre-sintered gelatin template by using both a centrifugation (strategy 2) and a vacuum (strategy 3) approaches. It is also important to point out that the use of reduced pressure in strategy 3 was possible because of the high boiling point of EL, equals to 150 °C, which limited solvent evaporation during the impregnation process.
Starting from these results, we have further improved the fabrication process previously described for the preparation of porous PLA–HA composite scaffolds. In Fig. 7 the results achieved in the case of strategies 1 and 3 are compared. Scaffolds prepared following strategy 2 easily disaggregated during handling, probably because of the incomplete infiltration of the polymeric solution. This effect can be explained by considering that the addition of HA accelerated the gelification of the polymeric solution. As a direct consequence, the room temperature centrifugation applied to strategy 2 induced a too fast increase of the viscosity of the PLA–HA/EL solution, making difficult the diffusion inside the void space of the gelatin template. Conversely, as shown in Fig. 7, both strategies 1 and 3 allowed for the preparation of porous PLA–HA composite scaffolds with a macroporous structure and a nanometre-scale fibrous architecture. It is also important to point out that, as observed in the case of neat PLA scaffolds, the use of a pre-sintered gelatin template allowed increasing the interconnectivity of the large pores. Close observation of the large-pore wall characteristics, reported in the insets of Fig. 7, evidenced that in the case of the PLA–HA composite scaffolds, the inorganic particles are quite uniformly distributed onto the surface of the pores.
 | ||
| Fig. 7 SEM images showing the morphology of PLA–HA composite scaffolds prepared by using 200–400 μm size gelatin particles as a function of HA concentration and obtained following strategies 1 and 3. | ||
This result was corroborated by those of EDX analysis reported in Fig. 8. As shown, red and green spots, which corresponded to the calcium and phosphate elements, respectively, are quite homogeneously distributed onto the pore surface, while the extent of detection of these two elements increased with the increase of the amount of HA in the starting solution. The EDX pattern reported in the inset of Fig. 8D evidenced the Ca and P peaks finally corroborating the presence of the inorganic particles onto the pores of the scaffolds.
 | ||
| Fig. 8 Results of SEM-EDX analysis carried out on PLA–HA composite scaffolds prepared following strategy 1 and showing the calcium (red spots) and phosphate (green spots) onto the surface of the pores of the scaffold. PLA–HA composite scaffold with (A and B) 10 wt% and (C and D) 30 wt% of inorganic filler. | ||
Porosity and pore size data of the PLA–HA scaffolds are reported in Fig. 9. Porosity values followed similar trends to those observed in the case of neat PLA scaffolds and evidenced the increase of scaffolds porosity from strategies 1 to 3. At the same time, the mean pore size increased with the increase of the size range of gelatin particles from 200–400 to 400–600 μm. Minor differences were observed on the porosity and large pores mean size as a function of the HA concentration in the scaffolds.
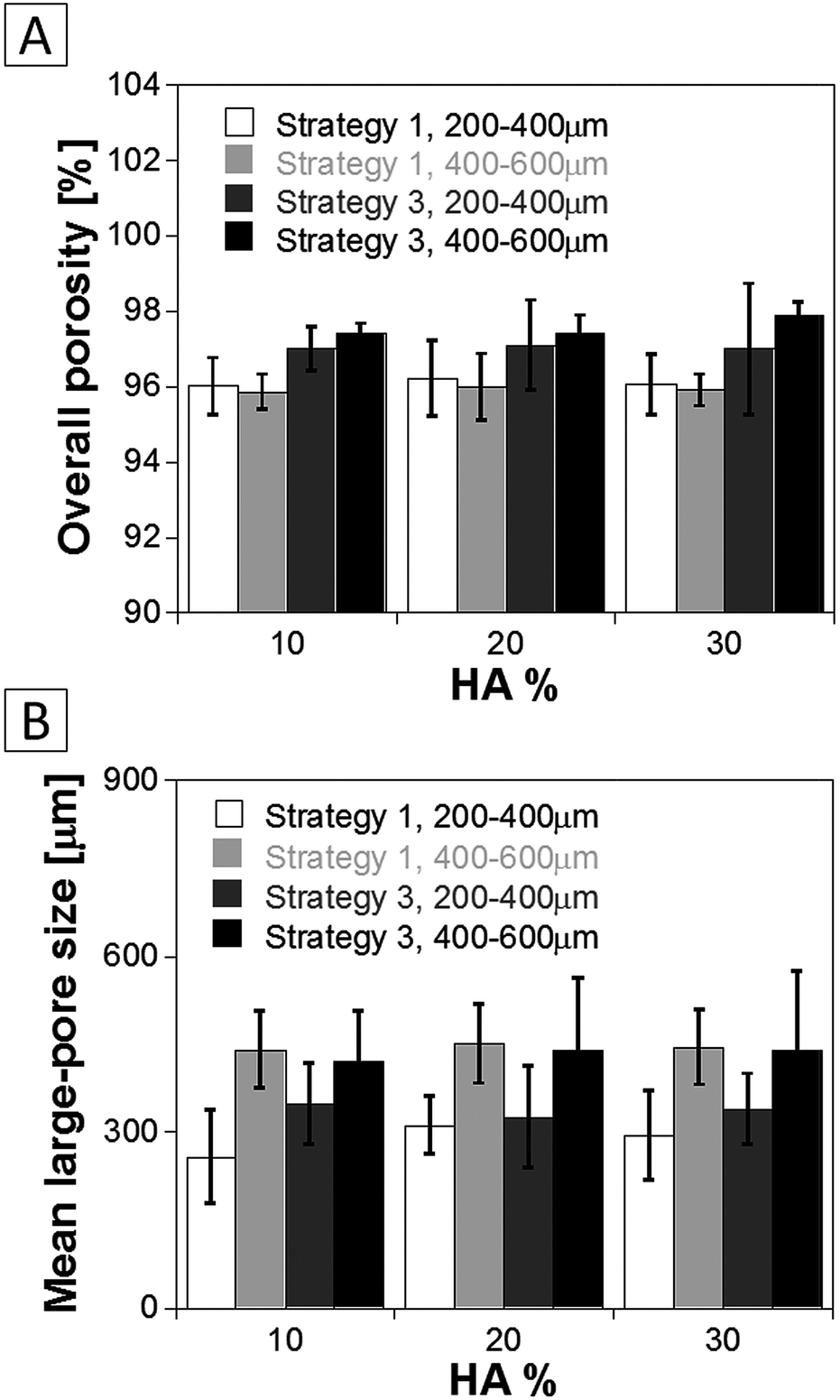 | ||
| Fig. 9 Porosity and mean size of large-pores of PLA–HA composite scaffolds prepared by means of strategies 1 and 3 as a function of the concentration of inorganic filler. | ||
Fig. 10 reports the results of the low temperature N2 adsorption–desorption analysis, which has been used to assess the nano scale pore structure and textural characteristics of the scaffolds as a function of their composition and manufacturing route. The pore volume distribution trends obtained by the analysis of the desorption curves are shown in Fig. 10A. As shown, the scaffolds have both mesopores and micropores and the pore volume fraction corresponding to these pores increase with the addition of HA nano-particles. The mean nano pores size and specific surface area results are reported in Fig. 11B. The mean nano-pore size of the PLA scaffold was close to 10 nm and decreased down to 7 nm after the addition of the HA filler. Concomitantly, the specific surface increases from 42 up to 123 m2 g−1 when 30% of HA was incorporated into the PLA scaffold. The PLA scaffolds prepared following strategy 3 evidenced a specific surface area value close to the scaffolds prepared following strategy 1. These results demonstrated that the synergy between the nano size scale fibres and the HA nanoparticles allowed achieving composite scaffolds with a high specific surface area and a nano porous pore wall texture.
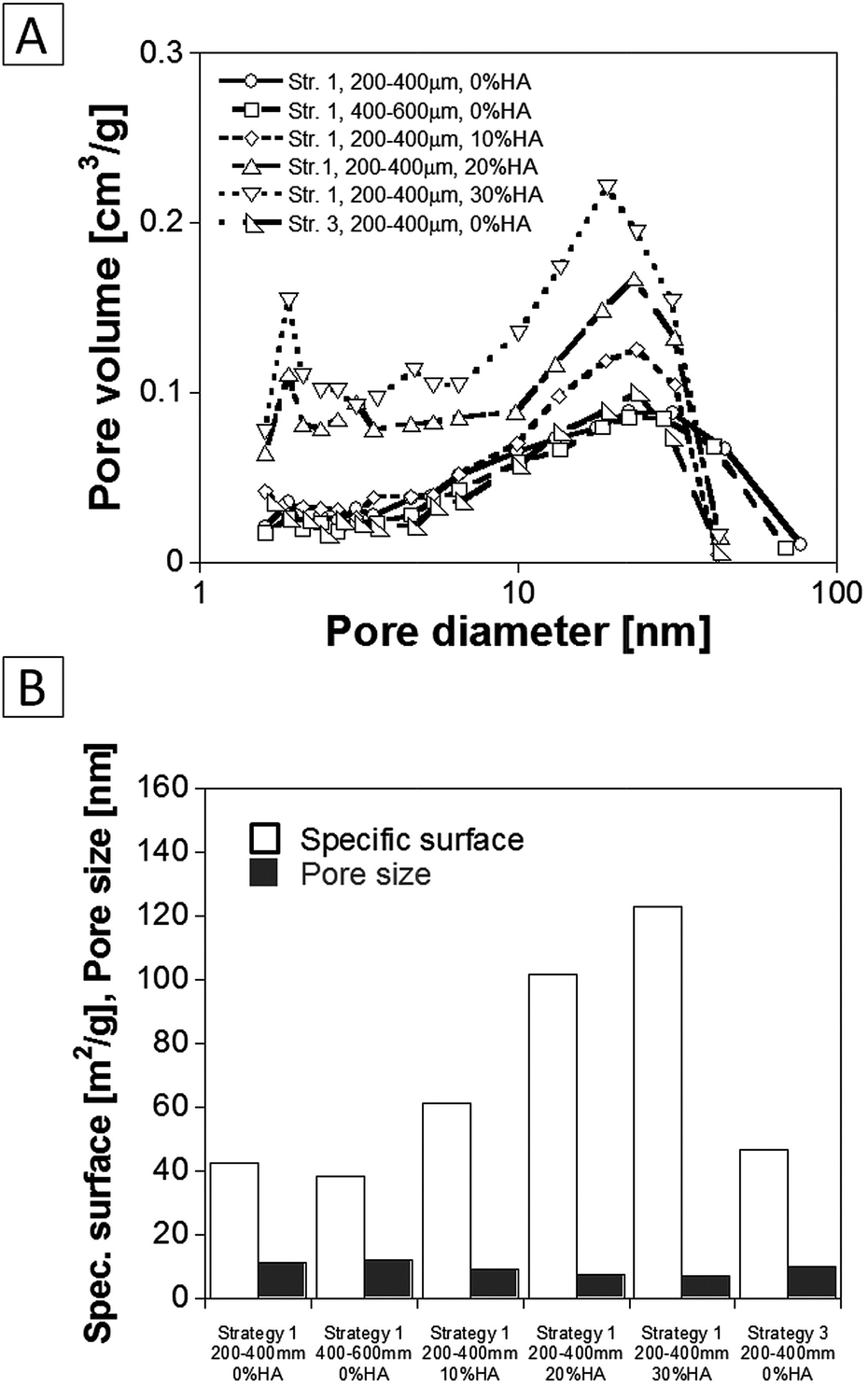 | ||
| Fig. 10 Results of BET analysis showing the textural features of the scaffolds as a function of their composition and structure: (A) nano-pore size distribution; (B) specific surface and mean nano-pore size. | ||
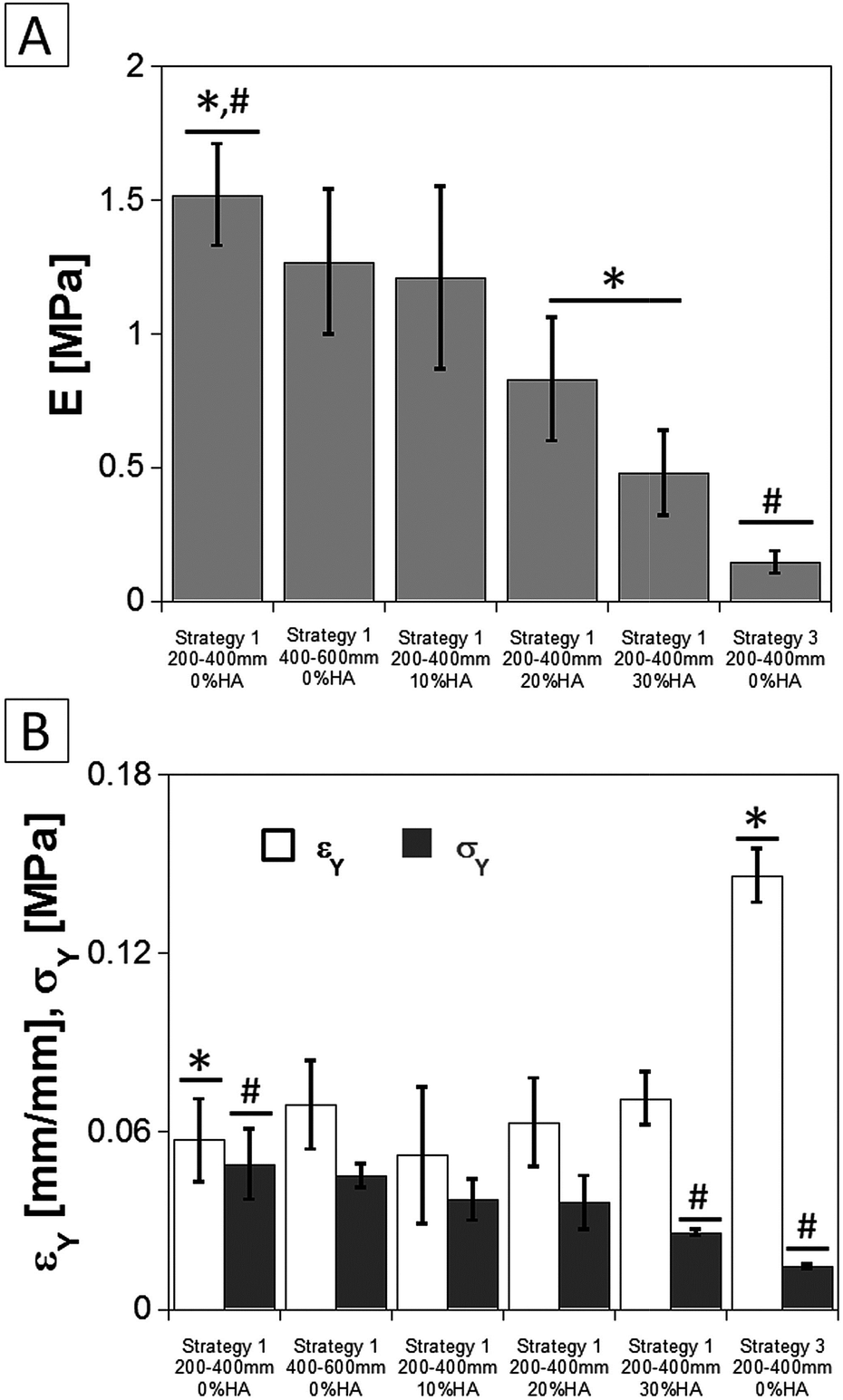 | ||
| Fig. 11 Mechanical properties of the scaffolds as a function of their composition and structure: (A) elastic modulus E; (B) compression yield strength (σY) and strain (εY). | ||
The presence of an interconnected porosity is an essential requirement in porous scaffolds for the easy adhesion and colonization of cells, as well as for the transport of fluids necessary for cell survival and growth. In this work, the percentage of interconnected porosity in the different scaffolds was calculated by assessing the volume percent of retained water inside the pores of the samples. The results of these tests (not shown) support the consideration that, independently of the composition and strategy used, the scaffolds were provided of a percent of interconnected open porosity in the 96–98% range.
The design of porous scaffolds with controlled mechanical properties is strongly required to promote cell adhesion and biosynthesis and, concomitantly, to define suitable in vitro and/or in vivo applications.1,3,4 Fig. 11 reports the values of the elastic modulus (E) and the compression yield strength (σY) and the strain (εY) of the scaffolds as a function of their composition and manufacturing route. The PLA scaffold prepared by using 200–400 μm gelatin particles and following strategy 1 was provided of an E value equal to 1.5 MPa. This value slightly decreased with the increase of the size range of gelatin particles. A more marked decrease of E was observed after the incorporation of the HA into the polymeric matrix. This effect was particularly evident for the samples containing 30 wt% of HA which was provided for an E value of 0.5 MPa, being it ascribable to the low chemical affinity between polymer and inorganic filler.30 The E value decreased further, down to 0.15 MPa, when PLA scaffolds were prepared using 200–400 μm gelatin particles following strategy 3. This effect was in agreement to the morphological characterization of the scaffolds, reported in Fig. 4, as the strategy 3 enhanced the interconnectivity between the large pores and reduced the pore wall thickness. The values of σY and εY corroborated the decrease of the mechanical response of the scaffolds after the incorporation of the HA as well as the lower stiffness of the scaffolds prepared by means of strategy 3.
The results of the in vitro cell–scaffold interaction study are reported in Fig. 12. As shown, the intensity of the Alamar Blue fluorescence, which is directly correlated to the number of viable cells in the scaffolds, increased with the culture time for all of the different scaffolds prepared. At day 1, the fluorescence intensity of control (CTR) was slightly higher than PLA and PLA–HA composite scaffolds, indicating enhanced cell adhesion onto the fibrin gel. The fluorescence intensity of the PLA scaffolds increased from about 2 × 103 at day 1 up to 9 × 103 at day 7 of culture. Minor differences were observed among the fluorescence intensity of the different scaffold types. It is important to point out that at day 7 the fluorescence intensity of the CTR was more than twice of that observed for the other samples, clearly indicating enhanced cell proliferation onto the fibrin gel.
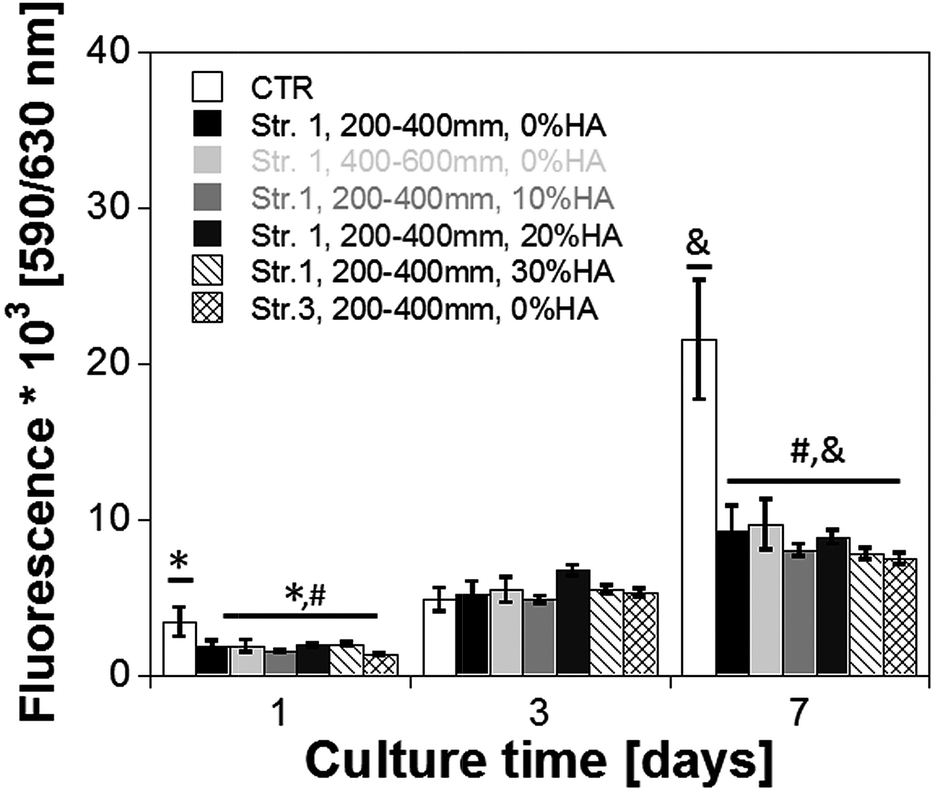 | ||
| Fig. 12 Osteoblast cells proliferation over 7 days of in vitro culture as assessed by means of Alamar Blue assay. | ||
Natural tissues, such as bone, are composed of a nano fibrous collagen matrix and a mineral phase consisting mainly of HA nano crystals. The possibility to replicate this peculiar composition and nano structure in de novo fabricated scaffolds is an important issue. Indeed, nano scale fibrous composite scaffolds can represent a step beyond traditional composite scaffolds, as they can combine the advantages of the chemical composition of polymer-composite materials to the “biomimetic” nature of the fibrous architecture, finally enhancing the biological response of the implant. Taken all together, the results of this study demonstrate that the developed bio safe process represents a powerful approach for the design and fabrication of porous PLA and PLA–HA composite scaffolds with potential application in TE. In particular, the process described in this work allows achieving biocompatible nano composite scaffolds with accurate control over morphology and textural features (e.g. pore size and interconnectivity), while reaching a high degree of porosity (up to 98%) and specific surface area (up to 123 m2 g−1). These characteristics are very difficult to achieve for nano composite scaffolds prepared by more traditional techniques, such as gas foaming or salt leaching. Furthermore, the results of the mechanical tests suggested that the proposed scaffolds can be especially useful for in vitro cell culture applications or soft tissue regeneration.
Conclusions
In this work, it is reported a novel bio safe process for the design and fabrication of porous PLA and PLA–HA composite scaffolds for TE. The process is based on the combination of TIPS, porogen leaching and scCO2 drying for the fabrication of porous biodegradable scaffolds with an interconnected network of large-pores and nanometre scale fibrous architecture.The optimization of the operating conditions, namely TIPS temperature, porogen size range and pre-treatment, as well as scCO2 drying conditions, with respect to different polymer–ceramic compositions is reported. In particular, we showed that the direct mixing of the polymeric solution and the porogen particles resulted in porous scaffolds with porosity up to 95%, while this procedure did not allow the control of the interconnection between large pores. Conversely, the use of a pre-sintered gelatin template enhanced both the porosity of the scaffolds, up to 97%, and the interconnectivity of the large pores. Nevertheless, when HA nanoparticles were used as the inorganic filler for scaffolds fabrication, the best scaffolds properties were achieved when the infiltration of the pre-sintered gelatin template with the polymeric solution was carried out following strategy 3.
Acknowledgements
Aurelio Salerno acknowledges the CSIC for the financial support through a JAE-DOC contract cofinancied by the FSE. The authors also gratefully acknowledge the financial support of the Ministerio de Economía y Competitividad through the research project BIOREG (MAT2012-35161). Additional support has been provided by the Generalitat of Catalonia under project 2014SGR-377.Notes and references
- A. Salerno and P. A. Netti, Biomedical Foams for Tissue Engineering, 2014, vol. 1, p. 3 Search PubMed.
- V. Liu Tsang and S. N. Bhatia, Adv. Drug. Deliv. Rev., 2004, 56, 1635 CrossRef CAS PubMed.
- P. X. Ma, Mater. Today, 2004, 7, 30 CrossRef CAS.
- V. Karangeorgiou and D. Kaplan, Biomaterials, 2005, 26, 5474 CrossRef PubMed.
- M. Martina and D. W. Hutmacher, Polym. Int., 2007, 56, 145 CrossRef CAS.
- A. Salerno, D. Guarnieri, M. Iannone, S. Zeppetelli, E. Di Maio, S. Iannace and P. A. Netti, Acta Biomater., 2009, 5, 1082 CrossRef CAS PubMed.
- P. A. Gunatillake and R. Adhikari, Eur. Cells Mater., 2003, 5, 1 CAS.
- J. R. Jones, Acta Biomater., 2013, 9, 4457 CrossRef CAS PubMed.
- A. Hoppe, N. S. Güldal and A. R. Boccaccini, Biomaterials, 2011, 32, 2757 CrossRef CAS PubMed.
- S. Bunting, L. Di Silvio, S. Deb and S. Hall, J. Hand Surg., 2005, 30, 242 CrossRef CAS PubMed.
- A. Salerno, M. Oliviero, E. Di Maio, P. A. Netti, C. Rofani, A. Colosimo, V. Guida, B. Dallapiccola, P. Palma, E. Procaccini, A. C. Berardi, F. Velardi, A. Teti and S. Iannace, J. Mater. Sci.: Mater. Med., 2010, 21, 2569 CrossRef CAS PubMed.
- K. Rezwan, Q. Z. Chen, J. J. Blaker and A. R. Boccaccini, Biomaterials, 2006, 27, 3413 CrossRef CAS PubMed.
- A. Salerno, S. Zeppetelli, E. Di Maio, S. Iannace and P. A. Netti, Compos. Sci. Technol., 2010, 70, 1838 CrossRef CAS PubMed.
- M. Yeo, H. Lee and G. Kim, Biomacromolecules, 2011, 12, 502 CrossRef CAS PubMed.
- F. Yang, R. Murugan, S. Ramakrishna, X. Wang, Y.-X. Ma and S. Wang, Biomaterials, 2004, 25, 1891 CrossRef CAS PubMed.
- J. Hu, X. Sun, H. Ma, C. Xie, Y. E. Chen and P. X. Ma, Biomaterials, 2010, 31, 7971 CrossRef CAS PubMed.
- V. La Carrubba, F. Carfi Pavia, V. Brucato and S. Piccarolo, International Journal of Material Forming, 2008, 1, 619 CrossRef PubMed.
- X. Liu and P. X. Ma, Biomaterials, 2009, 30, 4094 CrossRef CAS PubMed.
- A. Salerno and C. Domingo, Microporous Mesoporous Mater., 2014, 184, 162 CrossRef CAS PubMed.
- C. S. M. Pereira, V. M. T. M. Silva and A. E. Rodrigues, Green Chem., 2011, 13, 2658 RSC.
- L. Draghi, S. Resta, M. G. Pirozzolo and M. C. Tanzi, J. Mater. Sci.: Mater. Med., 2005, 16, 1093–1097 CrossRef CAS PubMed.
- X. Wang, G. Song and T. Lou, J. Mater. Sci.: Mater. Med., 2010, 21, 183 CrossRef CAS PubMed.
- A. Salerno, S. Zeppetelli, E. Di Maio, S. Iannace and P. A. Netti, Biotechnol. Bioeng., 2011, 108, 963 CrossRef CAS PubMed.
- G. B. A. Lim, S. S. Kim, Q. Ye, Y. F. Wang and D. R. Loyd, J. Membr. Sci., 1991, 64, 31 CrossRef CAS.
- N. Li, P. Luo, K. Liu, L. Chen, K. Wang, F. Chen and Q. Fu, J. Appl. Polym. Sci., 2011, 121, 604 CrossRef CAS.
- J. Shao, C. Chen, Y. Wang, X. Chen and C. Du, React. Funct. Polym., 2012, 72, 765 CrossRef CAS PubMed.
- W. L. Murphy, R. G. Dennis, J. L. Kileny and D. J. Mooney, Tissue Eng., 2002, 8, 43 CrossRef CAS PubMed.
- P. X. Ma and J. Choi, Tissue Eng., 2001, 7, 23 CrossRef CAS PubMed.
- M. Lebourg, R. S. Serra, J. M. Estellés, F. H. Sánchez, J. L. Gómez Ribelles and J. S. Antón, J. Mater. Sci.: Mater. Med., 2008, 19, 2047 CrossRef CAS PubMed.
- A. Salerno, M. Oliviero, E. Di Maio, P. A. Netti, C. Rofani, A. Colosimo, V. Guida, B. Dallapiccola, P. Palma, E. Procaccini, A. C. Berardi, F. Velardi, A. Teti and S. Iannace, J. Mater. Sci.: Mater. Med., 2010, 21, 2569 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |