
Alcohol mediated growth of α-MnO2 thin films from KMnO4 precursor for high performance supercapacitors
Abstract
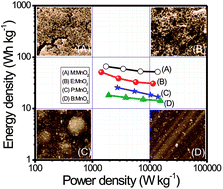
Energy storage devices, with low cost, high energy density, high power density, and long cycle life, have prime importance in order to solve the problem of interrupted power supply of renewable generation systems. In the present work, MnO2 thin films have been deposited by a simple, scalable, additive-free, binder-less, low cost, low temperature and eco-friendly chemical bath deposition method. The impact of three different alcohols (methanol, ethanol, 2-propenol) as reducing agents on the morphological, structural and electrochemical properties of MnO2 thin film is investigated. The MnO2 thin film prepared with the methanol as the reducing agent exhibits high specific surface area with excellent electrochemical features such as high specific capacitance of 633 F g−1 and high energy density of 65.9 W h kg−1 at current density of 1 mA cm−2 along with a good cycling stability of 95% after 2000 CV cycles. Such leading electrochemical properties suggest that MnO2 thin film prepared with methanol as the reducing agent using chemical bath deposition is a significant method to prepare reliable electrode material for future energy storage devices.
 Please wait while we load your content...
Please wait while we load your content...

