DOI:
10.1039/C4RA07684B
(Paper)
RSC Adv., 2014,
4, 45065-45073
Spectrofluorimetric determination of zearalenone using dispersive liquid–liquid microextraction coupled to micro-solid phase extraction onto magnetic nanoparticles†
Received
27th July 2014
, Accepted 5th September 2014
First published on 5th September 2014
Abstract
A new and sensitive method using dispersive liquid–liquid microextraction (DLLME) coupled to micro-solid phase extraction (μ-SPE) onto magnetic nanoparticles was developed for spectrofluorimetric determination of zearalenone (ZEN) in corn samples. In this study the solvent used to extract the analyte from a solid matrix, was then utilized as disperser solvent in the DLLME process. The DLLME was performed by injecting 3 mL of acetonitrile/water (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, v/v) (disperser) containing 320 μL of 1-heptanol (extraction solvent) into 30 mL of water sample. In the present DLLME-μ-SPE approach, hydrophobic magnetic nanoparticles were used to retrieve the extractant of 1-heptanol in the DLLME step. In fact the target of μ-SPE was the 1-heptanol rather than the ZEN. The ZEN was extracted from hydrophobic magnetic nanoparticles by stirring with 1 mL of acetonitrile for 4 min. Influential parameters affecting the extraction efficiency were investigated and optimized. Under the optimum conditions the calibration curve for ZEN determination showed good linearity in the range 0.51–300.0 μg L−1 (R2 = 0.9994) and the limit of detection (S/N = 3) was estimated to be 0.25 μg L−1. The intra-day and inter-day precision (RSD %) of ZEN were in the range of 2.7–4.1%. High recoveries ranging from 93.2 to 102.1% were obtained. The results demonstrated that the developed method is simple, inexpensive, accurate and remarkably free from interference effects. Also, this two-step method reclaimed the versatility of DLLME because the selection of the extraction solvent was not limited to the high density solvents.
:2, v/v) (disperser) containing 320 μL of 1-heptanol (extraction solvent) into 30 mL of water sample. In the present DLLME-μ-SPE approach, hydrophobic magnetic nanoparticles were used to retrieve the extractant of 1-heptanol in the DLLME step. In fact the target of μ-SPE was the 1-heptanol rather than the ZEN. The ZEN was extracted from hydrophobic magnetic nanoparticles by stirring with 1 mL of acetonitrile for 4 min. Influential parameters affecting the extraction efficiency were investigated and optimized. Under the optimum conditions the calibration curve for ZEN determination showed good linearity in the range 0.51–300.0 μg L−1 (R2 = 0.9994) and the limit of detection (S/N = 3) was estimated to be 0.25 μg L−1. The intra-day and inter-day precision (RSD %) of ZEN were in the range of 2.7–4.1%. High recoveries ranging from 93.2 to 102.1% were obtained. The results demonstrated that the developed method is simple, inexpensive, accurate and remarkably free from interference effects. Also, this two-step method reclaimed the versatility of DLLME because the selection of the extraction solvent was not limited to the high density solvents.
1. Introduction
Zearalenone (ZEN) is an estrogenic resorcylic acid lactone compound (Fig. 1) produced by Fusarium species, in particular Fusarium graminearum, Fusarium culmorum and Fusarium crookwellense that can infect and proliferate on various agricultural commodities in the field and/or during storage. ZEN occurs in mainly corn and wheat but it occurs also in barley, rice and sorghum amongst other food commodities frequently used in human and animal diets.1 ZEN is a strong estrogenic and anabolic compound that causes reproductive problems in farm animals. Symptoms may include vaginal swelling (vulvovaginitis) and, in severe cases, vaginal and rectal prolapse, especially in immature gilts (swine).2 Therefore, in order to minimize the risk to humans and animals, European Community legislation limits the concentration of ZEN for cereal-based foods intended for consumption by infants and young children at 20 μg kg−1 and for cereal products intended for adults at 100 μg kg−1.3 Several analytical methods have been reported for the determination of ZEN such as thin-layer chromatography (TLC),4 enzyme-linked immunosorbent assay (ELISA),5 high-performance liquid chromatography (HPLC),6–9 ultra-performance liquid chromatography with tandem mass spectrometry (UPLC-MS-MS),10 spectrofluorimetry with molecularly imprinted optosensing material (MIOM),11 fluorescence resonance energy transfer immunoassay (FRETI),12 and fluorescent-labeled immunosorbent assay (FLISA).13 Although some of these analytical techniques, such as HPLC and UPLC-MS-MS, benefit from high sensitivity and low detection limit, they require the involvement of skilled personnel and expensive instrumentation. The development of a new method with simplicity, reliability, high sensitivity and specificity for routine analysis of ZEN is desirable. Thus, spectrofluorimetry can be considered as a valuable method because of its simplicity, high sensitivity, relative selectivity, low cost, and less time consuming.14,15 Since the matrices of the food samples are often complex, determination of ZEN in real samples requires a pretreatment step for sample enrichment and clean-up before analysis. Generally pretreatment step involves an acetonitrile–water (8:2, v/v) extraction followed by a clean-up step. Various purification methods have been reported for extraction and clean-up of ZEN such as dispersive liquid–liquid microextraction (DLLME),16 solid phase extraction (SPE) with C18 cartridges,17 solid phase extraction with molecular imprinting polymer (MIP),6,7,11 and solid-phase extraction with immunoaffinity column (IAC).18 IAC is the most common clean-up method which allows a highly selective separation of analyte from a complex matrix.15 However, IAC has some important disadvantages such as relatively high cost, lake of reusability, long operation time and limited shelf-life.19,20 Recently, dispersive liquid–liquid microextraction (DLLME) has been introduced as a single step separation and preconcentration method and it has found extensive application, as highlighted by several reviews.21–24 Conventional DLLME is based on a ternary component solvent system in which an appropriate mixture of the extracting solvent and disperser solvent is rapidly injected into the aqueous sample. Then, the extracting solvent is dispersed into the aqueous phase and target analytes are extracted into the fine droplets of extracting solvent.21 Next, centrifugation is applied to sediment the extracting solvent from water samples. The extracting solvent containing the extracted analytes is then withdrawn by using a syringe and subjected to final analysis. Conventional DLLME was restricted to the usage of a high-density solvent. Generally, organic solvents denser than water are quite toxic and harmful to the environment. Also, the numbers of organic solvents denser than water are limited to chlorinated solvents such as chlorobenzene, chloroform, tetrachloromethane and tetrachloroethane. Then, the use of a solvent denser than water is a disadvantage and limits wide applicability of DLLME. In recent years, DLLME with a low-density organic solvent as the extractant had been developed to overcome these disadvantages.25,26 But it requires additional processing steps, apart from the mandatory centrifugation, including refrigeration to freeze the organic solvent, manually retrieving it to let it thaw, and use of additional materials such as surfactants or an apparatus such as special test tubes.25–27 To overcome these drawbacks, DLLME coupled with μ-SPE (DLLME-μ-SPE) has been introduced for the determination of some analytes such as metal chelates, polycyclic aromatic hydrocarbons and 4-n-nonylphenol.27–29 In this method, μ-SPE based on hydrophobic magnetic nanoparticles is applied to retrieve the extraction phase of DLLME by adsorption. Then, separation is quickly carried out by the application of an external magnetic field. This two-step microextraction procedure lacks tedious steps such as centrifugation, refrigeration to freeze and manual collection of extraction phase. The aim of this study was to investigate the applicability of the DLLME coupled with μ-SPE for enhanced spectrofluorimetric determination of ZEN in corn samples. 1-Heptanol was used as extraction solvent in DLLME step and dispersed as fine droplets. The formed micro-emulsion phase can retrieve ZEN and then is rapidly partitioned on the surface of hydrophobic magnetic nanoparticles. Since, 1-heptanol is a large alcohol with non-polar hydrophobic region, a hydrophobic interaction can be occurs between 1-heptanol and hydrophobic magnetic nanoparticles. To the best of our knowledge, this is the first report about application of DLLME-μ-SPE for separation and spectrofluorimetric determination of ZEN in corn samples. All the experimental parameters affecting the two-step extraction were investigated in details and the analytical characteristics of the method were evaluated. The method was demonstrated to be applicable for the analysis of ZEN in cereal samples.
 |
| | Fig. 1 The molecular structure of zearalenone. | |
2. Experimental
2.1. Standards and materials
The standard solution of ZEN (10000 μg L−1 in acetonitrile) and all HPLC-grade solvents such as acetone (Me2CO), acetonitrile (MeCN), dichloromethane (CH2Cl2), methanol (MeOH), ethanol (EtOH), ethyl acetate (C4H8O2), toluene (C6H5–CH3), 1-heptanol (C7H16O), 1-octanol (C8H18O), 2-ethylhexanol (C8H18O), diethyl ether ((C2H5)2O), 1,4 dioxane, and water (H2O) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Iron(III) chloride hexahydrate (FeCl3·6H2O), Iron(II) chloride tetrahydrate (FeCl2·4H2O), tetraethyl orthosilicate (TEOS) and other used chemicals were supplied by Merck (Darmstadt, Germany). As safety notes, all used laboratory glassware were treated with an aqueous solution of sodium hypochlorite (5%) before the discarding to minimize health risks due to ZEN contamination.
2.2. Instrumentation
The fluorescence measurements were performed using a Cary Eclipse Fluorescence Spectrophotometer (Varian, USA) equipped with a xenon lamp. All measurements were performed in 10 mm quartz microcells, at room temperature. Spectra recording were carried out in fluorescence scan mode with the slit widths of 5 nm. The PMT detector was used for recording the emission lines and set on 600 V. The modified magnetic nanoparticles were characterized by an H-800 transmission electron microscope (TEM) (Hitachi, Japan), APD2000 X-ray diffractometer (XRD) (Italstructures, Italy) and FT-IR spectrometer (Perkin Elmer, spectrum version 10.01.00, USA). A permanent magnet of Nd-Fe-B (100 mm × 50 mm × 40 mm, Model N48, China) was used for magnetic separation. Vortex mixer Model L46 (LABIN Co., Netherlands) was used for better combining and accelerating reaction between reagent.
2.3. Synthesis of TEOS functionalized magnetic nanoparticles
The magnetic nanoparticles (MNPs) were prepared via improved chemical co-precipitation method. FeCl3·6H2O (11.68 g) and FeCl2·4H2O (4.30 g) were dissolved in 200 mL deionized water under nitrogen atmosphere with vigorous stirring at 85 °C. Then, 20 mL of 30% aqueous ammonia solution was added to the solution. The color of the bulk solution changed from orange to black immediately. The magnetic precipitate was washed twice with deionized water and once with 0.02 mol L−1 sodium chloride solution.30,31 The washed MNPs were stored in deionized water at a concentration of 40 g L−1. Then, 20 mL of above prepared magnetic suspension was placed in a 250 mL round-bottom flask and allowed to settle. The supernatant was removed and coating of MNPs with TEOS was carried with the addition of an aqueous solution of TEOS (10%, v/v, 80 mL), followed by glycerol (60 mL). The mixture was then stirred and heated at 90 °C for 2 h under a nitrogen atmosphere. After that, the resulting modified nanoparticles (TEOS–Fe3O4) were washed with deionized water (3 × 250 mL), methanol (2 × 150 mL), deionized water (3 × 250 mL) and dried as black powders in a vacuum oven at 45 °C for 2 h.32
2.4. Real sample pretreatment
Corn samples were purchased from a local market and were stored at 4 °C until their analysis. These samples were weighed and 25 g of thoroughly homogenized were extracted with 100 mL of a mixture of MeCN/H2O (8:2, v/v) with a blender at high speed for 3 min. The extracts were filtered on a filter paper (Whatman No. 44) and then processed by DLLME.
2.5. Analytical procedure
320 μL of 1-heptanol (as extraction solvent of DLLME) was added to an aliquot of 3 mL of MeCN 80% extract (used as disperser solvent) and the mixture was rapidly injected into a 30 mL vial with conical bottom containing 15 mL of water. Then, the vial was sealed and swirled on a vortex agitator at 3500 rpm for 1 min (equilibration time). After that, 50 mg of the magnetic nanoparticles were quickly added to the vial. The solution was stirred for 3 min to facilitate adsorption of target analyte on the surface of MNPs. Then, the magnetic adsorbent was collected using an external magnet and supernatant water was decanted. The adsorbed ZEN was desorbed from surface of the adsorbent by the addition of 1 mL MeCN and stirring for 4 min. Finally, the magnet was used again to settle the nanoparticles, and the desorbed solution was evaporated under a gentle nitrogen flow. The residue was reconstituted in 300 μL of diethyl ether for spectrofluorimetric detection.
3. Results and discussion
The analysis of low levels of contaminants in solid matrices such as foods and food products requires a sample treatment, before analysis and a purification procedure of the extract to increase sensitivity and achieve low levels of detection. In this study, DLLME-μ-SPE was used as a clean-up and preconcentration technique of solid sample extract and spectrofluorimetry has been applied for determination of ZEN. The intensity of the fluorescence peak was used to assess the extraction efficiency under various conditions (the wavelengths of 270 and 380 nm were used as maximum excitation and emission wavelengths). A univariate approach was employed to optimize influential factors in this method and all results were average of three replicate measurements.
3.1. Characterization of the adsorbent
To confirm that TEOS is bonded to the Fe3O4 NPs, the characterization was performed by FT-IR spectroscopy. The FT-IR spectra for Fe3O4 and TEOS–Fe3O4 are shown in Fig. 2a and b. The characteristic peak of Fe3O4 nanoparticles can be seen in Fig. 2a, as a strong absorption band at 571 cm−1 which corresponds to Fe–O band of bulk magnetite. This band can be observed in TEOS–Fe3O4 spectrum too. The broad feature in the range 3441–3220 cm−1 is due to O–H stretching vibration, which corresponds to the hydroxyl groups attached by the hydrogen bonds to the iron oxide surface (Fig. 2a). After initial coating step, the characteristic peaks at 1103–1030 cm−1 are related to the O–Si stretching vibration (Fig. 2b). Also Fig. 3a displays the TEM image of TEOS–Fe3O4, which illustrates the relatively uniform size distribution of this adsorbent with a mean diameter of approximately 10 ± 1.2 nm. X-ray diffraction patterns of TEOS-MNPs was shown in Fig. 3b, representing the reflection patterns at peak position (2θ) of about 30.2, 35.3, 43.2, 57.2, 62.7, and 74.2 which correspond to the reflection planes of 220, 311, 400, 511, 440, and 622, respectively. The position and relative intensity of all diffraction peaks are consistent with the standard pattern of Fe3O4 according to the JCPDS card.33 The average particle size of TEOS–Fe3O4 adsorbent using on the Scherrer equation based on the most intense XRD peak (311-diffraction peak, 2θ = 35.3) was calculated 9.5 nm which is in good agreement with that obtained of used TEM image.
 |
| | Fig. 2 FT-IR spectra of MNPs (a) and TEOS-MNPs (b). | |
 |
| | Fig. 3 SEM image of TEOS-MNPs (a) and X-ray diffraction pattern of TEOS-MNPs (b). | |
3.2. Optimization of the DLLME-μ-SPE method
3.2.1. Selection of the disperser solvent. The solvent used to primary extract of the analytes from solid matrix must then act as disperser solvent in DLLME process, therefore, its selection must take into account both the properties required to the primary extracting solvent and DLLME dispersant.34 Generally, an aqueous mixture of MeCN (MeCN 80%) was applied for the extraction of ZEN from food samples,6,11 while Me2CO, MeCN and MeOH are usually used as disperser solvents in DLLME method. On the basis of these considerations, the usefulness of several solvents, including Me2CO, MeOH, MeCN, EtOH, MeOH 80% and MeCN 80% was investigated in the preliminary experiments. The extraction efficiencies achieved with MeCN 80% were higher than other solvents (see Fig. 4). Therefore MeCN 80% was selected as extraction solvent of ZEN from the cereal samples and as disperser solvent in DLLME for subsequent experiments. Furthermore, the effect of disperser solvent volume on ZEN recovery was investigated in the range of 1–5 mL. The obtained results (Fig. S1, ESI†) showed that the extraction efficiency increased with increasing volume of MeCN 80% to 3 mL and then decreased at higher volumes due to the increased solubility of ZEN in the aqueous phase. Also, this led to a decrease in extraction efficiency because of a decrease in the distribution ratio. Based on the obtained results, further studies were performed with 3 mL of MeCN 80%.
 |
| | Fig. 4 Effect of dispersive solvent type. Conditions: dispersive solvent volume, 4 mL containing 5 μg L−1 of ZEN; extracting solvent volume and type, 310 μL of 1-heptanol; water volume, 15 mL, equilibration time, 120 s, adsorbent amount, 80 mg; adsorption time, 5 min; desorption time, 5 min, desorption solvent volume and type, 1 mL of MeCN; reconstituting solvent, 300 μL of diethyl ether; without salt addition. Error bars represent the standard deviation for three experiments. | |
3.2.2. Optimization of DLLME. The effect of various experimental parameters, such as the type and the volume of the extraction solvent, salt addition, equilibration time and water volume were investigated. The selection of a suitable extracting solvent is of great importance for the optimization of DLLME process. Also, for new DLLME method an extracting solvent must have several characteristics: it should have good emulsification efficiency in the aqueous sample, high affinity for compounds of interest, low solubility in water, low density and a low vapor pressure to prevent loss during agitation. On the basis of these considerations, the usefulness of several low-density organic solvents, including ethyl acetate, toluene, 1-heptanol, 1-octanol and 2-ethylhexanol were investigated in the preliminary experiments. Among them a stable cloudy solution and good extraction efficiency were observed with 1-heptanol (Fig. 5). The volume of extracting solvent is an important parameter which can influence the occurrence of the cloudy state and efficiency of extraction process. The effect of extracting solvent volume on the extraction of ZEN was investigated in the range of 250–350 μL. The results are shown in Fig. S2.† As can be seen, fluorescence intensity of ZEN increased with increasing the volume of extracting solvent from up to 310 μL and then decreased with further increases in solvent volume due to dilution effects. The volumes smaller than 250 μL were avoided due to dissolution of organic solvent in aqueous phase. Therefore, the volume of 320 μL was selected as an optimum solvent volume for further studies. Addition of the salt to the sample may have several effects on the extraction efficiency. Generally, the addition of salt can decrease the solubility of target analytes in the aqueous phase and promote the transfer of analytes toward the organic phase and thus improve the extraction efficiency (salting-out).21 Also addition of the salt increases the viscosity and density of the solution. This can reduce the efficiency of emulsification phenomenon because lower solubility of extracting solvent in aqueous phase. In this study, the effect of salt addition on the extraction efficiency was investigated by addition of different amounts of NaCl (0–5% w/v) into the spiked samples. The results were shown that the extraction efficiency of ZEN was almost constant in the range of 0–5% (Fig. S3†). Then, no addition of salt was chosen in the subsequent experiments. The effect of water volume on the ZEN extraction was investigated using different water volumes in the range of 3–25 mL. The rustles were shown that the analyte recoveries were also affected statistically by water volume and to obtain a higher enrichment factor, a larger volume of water is required. On the other hand, the extraction efficiency would decrease at very high water volumes due to the increased solubility of ZEN in the aqueous phase. The extraction efficiency was constant in the range of 3–18 mL and then decreased at higher water volumes (Fig. S4†). Thus, the volume of 15 mL of water was selected for subsequent experiments. The last factor of the DLLME step was the equilibration time which is important in the most microextraction procedures. In this work, equilibration time is defined as interval time from the occurrence of the cloudy state and just before addition of the hydrophobic magnetic nanoparticles. Equilibration time was investigated in the range of 0–300 s maintaining the rotational speed at the maximum level (3500 rpm) to maximize energy transfer and reduce mixing time. Results (Fig. S5†) indicated that fluorescence intensity increased with increasing of equilibration time up to 60 s and then levelled off with further increases in time. Thus the minimum time of 60 s was selected as equilibration time for subsequent experiments.
 |
| | Fig. 5 Effect of extracting solvent type. Conditions: dispersive solvent volume and type, 3 mL of MeCN 80% containing 5 μg L−1 of ZEN; extraction solvent volume, 310 μL; water volume, 15 mL, equilibration time, 120 s, adsorbent amount, 80 mg; adsorption time, 5 min; desorption time, 5 min, desorption solvent volume and type, 1 mL of MeCN; reconstituting solvent, 300 μL of diethyl ether; without salt addition. Error bars represent the standard deviation for three experiments. | |
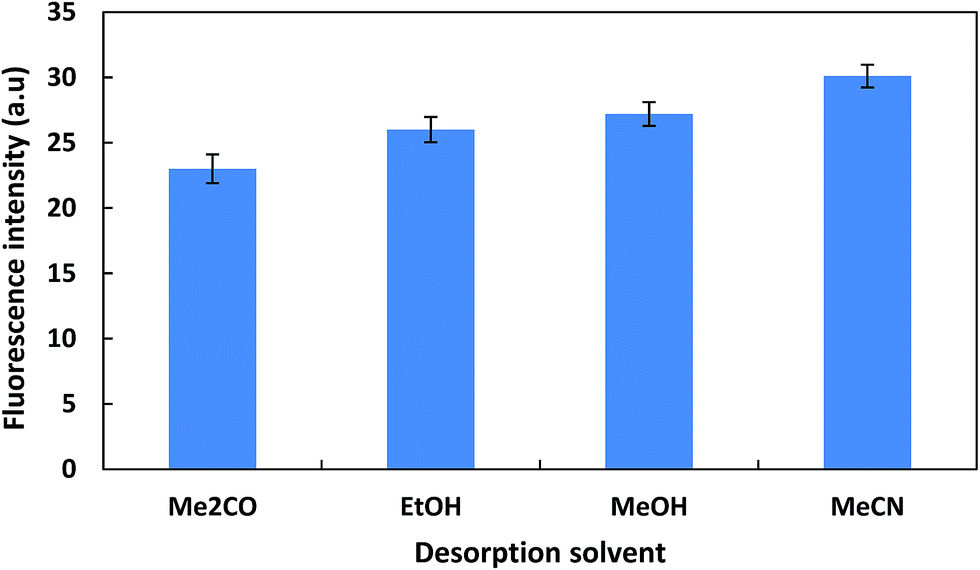
3.2.3. Optimization of magnetic μ-SPE step. The parameters associated with the magnetic μ-SPE step, involving the amount of hydrophobic MNPs (TEOS–Fe3O4), adsorption and desorption times, type and volume of desorption solvent, were investigated and optimized. The amount of hydrophobic MNPs (TEOS–Fe3O4) is important parameter to accomplish quantitative removal of the extraction phase, containing the ZEN. Then, the different amounts of TEOS–Fe3O4 were investigated in the range 10–100 mg. The results showed that the extraction efficiency increased with increasing amounts of adsorbent up to 70 mg and then leveled off (Fig. S6†). Therefore, 50 mg of TEOS–Fe3O4 was selected for the further experiments. For studying the effect of adsorption time on extraction efficiency, adsorption time was investigated in the range of 1–10 min and obtained results showed that an adsorption time of 3 min was sufficient to attain adsorption equilibrium (Fig. S7†). Afterwards, the usefulness of several of organic solvents as desorption solvent was investigated in desorption step (Fig. 6). As can be seen the best result was found with 1 mL of MeCN. The effect of desorption solvent volume on ZEN recovery was further investigated in the range of 0.3–2 mL and the maximum sensitivity was obtained over the range 0.8–2 mL (Fig. S8†). Therefore, 1 mL of acetonitrile was selected. Also the effect of desorption time was investigated in the range of 1–7 min (Fig. S9†). A duration time of 4 min appeared to be sufficient for complete desorption.
 |
| | Fig. 6 Effect of desorption solvent type. Conditions: dispersive solvent volume and type, 3 mL of MeCN 80% containing 5 μg L−1 of ZEN, extracting solvent volume and type, 320 μL of 1-heptanol, water volume, 15 mL, equilibration time, 60 s, adsorbent amount, 50 mg; adsorption time, 3 min; desorption time, 5 min, desorption solvent volume, 1 mL; reconstituting solvent, 300 μL of diethyl ether; without salt addition. Error bars represent the standard deviation for three experiments. | |
3.3. Reconstituting solvent effect
Solvent polarity has a remarkable effect on the fluorescence intensity of ZEN. The fluorescence intensity of ZEN increases with reducing of solvent polarity, a property known to influence fluorescence properties.35,36 The influence of polarity of solvent on the fluorescence of ZEN was examined by investigating the effect of several organic solvents such as acetonitrile, acetone, methanol, ethanol, diethyl ether and 1,4 dioxane on fluorescence intensity (Fig. S10†). The experimental results showed that the greatest enhancement was observed in diethyl ether. Therefore, to enhance the fluorescence efficiency of ZEN, desorbing solvent was evaporated and residual was reconstituted in 300 μL of diethyl ether.
3.4. Analytical parameters
Under the selected experimental conditions, a linear calibration graph based on series standards was obtained over the range 0.51–300.0 μg L−1 of ZEN standard solutions with the linear regression equation If = 4.4819C + 8.1858 (If, fluorescence intensity and C, μg L−1 of ZEN) and correlation coefficient R2 = 0.9994. Limit of detection (LOD = 3.3Sb/m, where Sb is the standard deviation for ten blank measurements and m is the slope of the calibration curve) was found to be 0.25 μg L−1. The precision of the method was evaluated (as RSD %) through investigation intra-day precision and inter-day precision. The intra-day precision was evaluated over five replicates spiked at two concentration levels (1 and 5 μg L−1 of ZEN) within one day (n = 5). The inter-day precision was evaluated over five daily replicates, spiked at same level per work day, over a period of three days (n = 15). The results were listed in Table 1. Furthermore, to investigate the possible matrix effect on the ZEN determination in real sample, the limits of matrix-matched detection (LOD, S/N = 3) and quantification (LOQ, S/N = 10) were evaluated from matrix-matched calibration. The values of MM-LOD and MM-LOQ were obtained to be 0.28 μg kg−1 and 0.58 μg kg−1, respectively. Solutions for matrix-matched calibration were prepared by spiking appropriate amounts of ZEN working solutions to the none-contaminated corn sample and following the DLLME-μ-SPE procedure and fluorescence measurement. The results indicated that sample matrix cannot significantly affect the ZEN determination. Also, enrichment factor (EF) was calculated by EF = VS/VR × R% definition (where VS is the sample volume, VR is the reconstituting solvent volume, and R% is extraction yield). In this study, by extracting 18 mL of sample solution into 300 μL of reconstituting solvent (recovery = 97.5%), the enrichment factor of 58.5 was achieved for ZEN determination by the developed method. Adsorption capacity of adsorbent is investigated by static desorption method. For this purpose 50 mg of hydrophobic adsorbent was equilibrated with 18 mL of dispersed analyte solution after DLLME step, containing various concentrations at optimum conditions. After 10 mine the mixture was filtered and supernatant were analyzed. The results showed that the amount of analyte adsorbed per unit mass of adsorbent was increased linearly with the initial concentration of ZEN and then was reached to a plateau value (adsorption capacity value), which represent saturation of the active surface of hydrophobic adsorbent for ZEN. The maximum adsorption capacity of prepared adsorbent for ZEN was found to be 0.625 mg g−1.
Table 1 The characteristic data of the proposed method
| Parameters |
Data |
| For 1 μg L−1 of ZEN. For 5 μg L−1 of ZEN. Sb is the standard deviation for ten blank measurements and m is the slope of the calibration curve. |
| Dynamic range (μg L−1) |
0.5–300 |
| Correlation coefficient (R2) |
0.9994 |
| Intra-day precision (RSD %, n = 5) |
3.6a |
| 2.7b |
| Inter-day precision (RSD %, n = 15) |
4.1a |
| 3.1b |
| Limit of detection (3.3Sb/mc, μg L−1) |
0.25 |
3.5. Selectivity study
Selectivity and competitive extraction experiments were carried out using zearalenone (ZEN), aflatoxins (AFB1, AFB2, AFG1 and AFG2), ochratoxin A (OTA) and deoxynivalenol (DON) which are other mycotoxins that may exist in cereals. Therefore, the possible interference effects of total AFs, DON and OTA was studied by co-existing of them alone and in mixture. The obtained results (Table 2) showed that the recoveries were not significantly affected by the presence of the interferences, indicating good selectivity for determination of ZEN in corn.
Table 2 Effect of mycotoxins interferences on the extraction efficiency of ZEN (5 μg kg−1)
| Interferences |
Concentration (μg kg−1) |
Recovery ± RSD (%) |
| Aflatoxins |
5 |
94.9 ± 2.7 |
| OTA |
5 |
96.3 ± 2.6 |
| DON |
5 |
95.2 ± 3.1 |
| Mixture |
Total |
94.1 ± 2.5 |
3.6. Real sample analysis
To test the applicability of the proposed method in real cereal samples, it was applied to the determination of ZEN in corn samples. Recovery studies were carried out by spiking the samples with different amounts of ZEN. Results (Table 3) showed that the recovery values were in the range of 93.4 to 103.1%. Also, Fig. 7 shows the typical spectra of the spiked (5 μg kg−1 of ZEN) and non-spiked corn sample at optimum working conditions. Comparison of the spectra and acceptable recoveries demonstrated that the matrices of corn sample had no effects on the performance of the presented method. Accuracy of the developed method for the determination of ZEN in two contaminated real samples was checked with IAC-HPLC-FD results (the AOAC standard method).37 The results are presented in Table 4. The statistical analysis of the results using Student's t-test showed that there are no significant differences between results obtained by two methods at 95% confidence level. Furthermore a comparison of the analytical feature achieved by the proposed method and other methods for ZEN determination is presented in Table 5. The presented method has distinct advantages in term of low detection limit, wide linear range, ease of operation and simplicity.
Table 3 Determination of ZEN in spiked corn samples
| Corn sample |
Spiked (μg kg−1) |
Founda (μg kg−1) |
Recovery (%) |
RSD % |
| Mean of three determinations. ND, not detected. |
| Sample 1 |
0.00 |
NDb |
— |
|
| 10.00 |
9.68 |
96.8 |
2.4 |
| 15.00 |
14.31 |
95.4 |
2.1 |
| 20.00 |
20.41 |
102.1 |
1.9 |
| Sample 2 |
0.000 |
ND |
— |
|
| 10.00 |
9.34 |
93.4 |
2.6 |
| 15.00 |
14.56 |
97.1 |
2.2 |
| 20.00 |
18.86 |
94.3 |
1.8 |
| Sample 3 |
0.000 |
ND |
— |
|
| 10.00 |
9.85 |
98.5 |
2.5 |
| 15.00 |
15.23 |
101.5 |
2.0 |
| 20.00 |
18.64 |
93.2 |
1.7 |
 |
| | Fig. 7 The typical spectra of non-spiked corn (blank) (a) and spiked corn (b). Conditions: dispersive solvent volume and type, 3 mL of MeCN 80% containing 5 μg L−1 of ZEN; extracting solvent volume and type, 320 μL of 1-heptanol; water volume, 15 mL; equilibration time, 60 s; adsorbent amount, 50 mg; adsorption time, 3 min; desorption time, 4 min; desorption solvent volume and type, 1 mL of MeCN; reconstituting solvent, 300 μL of diethyl ether; without salt addition. Error bars represent the standard deviation for three experiments. | |
Table 4 Comparison of ZEN analyses (mean ± SD, n = 3) in contaminated corn samples by proposed method and HPLC-FD method
| Corn sample. |
Proposed method |
HPLC-FDa |
| ZEN (μg kg−1) |
ZEN (μg kg−1) |
| HPLC analysis by AOAC standard method.37 |
| Sample 1 |
2.51 ± 0.07 |
2.66 ± 0.08 |
| Sample 2 |
10.34 ± 0.23 |
10.11 ± 0.27 |
Table 5 Comparison of diverse methods for the determination of ZEN
| Method |
Matrix |
LOD (μg kg−1) |
Linear range (μg kg−1) |
Recovery (%) |
Reference |
| Quick Easy Cheap Effective Rugged and Safe method. |
| QuEChERSa-HPLC-LSD |
Barley |
1.56 |
0.1–10 |
83.6–91.5 |
2 |
| MIP-SPE-HPLC-FD |
Corn, wheat |
— |
20–8800 |
82–87 |
6 |
| MIP-SPE-HPLC-FD |
Wheat, barley, corn, |
1.7–2.4 |
6–500 |
86–97 |
7 |
| IAC-HPLC-FD |
Wheat, barley, maize |
3.5–17.6 |
— |
84.0–105.0 |
8 |
| SPE-HPLC-DAD |
Corn |
0.7 |
0–400 |
90.0 |
9 |
| DLLME-μ-SPE-Spectrofluorimetry |
Corn |
0.58 |
0.51–300 |
93.2–102.1 |
This work |
4. Conclusion
A new two-step microextraction procedure, based on DLLME coupled with μ-SPE with hydrophobic magnetic nanoparticles, was developed for spectrofluorimetric determination of zearalenone in corn samples. In this method, DLLME is directly used for extraction and separation of ZEN from solid matrix and μ-SPE is applied to collect the extraction phase of DLLME. The developed method lacks tedious steps of conventional microextraction methods, such as centrifugation, refrigeration and thawing of organic solvent and manual collection of extraction phase, and is fast. Also, it is demonstrated that an organic solvent with lower density than water can be used in DLLME without involving any special apparatus. Other advantages of this method are simplicity of the extraction, minimum organic solvent consumption, excellent enrichment in a short extraction time, good repeatability and reproducibility, low cost and high accuracy. The good spiked recoveries of ZEN in real samples and the inherent high sensitivity and selectivity of spectrofluorimetric method showed that the present method was sufficiently applicable for determination of ZEN in real samples.
References
- International Agency for Research on Cancer (IARC), World Health Organization (WHO), Lyon, France, 1993, vol. 56, pp. 397–404.
- J. Wu, R. Zhao, B. Chen and M. Yang, Food Chem., 2011, 126, 1508–1511 CrossRef CAS PubMed.
- European Commission regulation (EC). No. 1126/2007, Off., J. Eur. Union, 2007, L. 255, pp. 14–17.
- B. L. Sekiyama, A. B. Ribeiro, P. A. Machinski and M. Machinski Jr, Braz. J. Microbiol., 2005, 36, 289–294 CrossRef CAS PubMed.
- Y. Liu, C.-zh. Zhang, X.-y. Yu, Zh.-y. Zhang, X. Zhang, R.-r. Liu, X.-j. Liu and Zh.-m. Gong, J. Zhejiang Univ., Sci., B, 2007, 8, 900–905 CrossRef CAS PubMed.
- P. Lucci, D. Derrien, F. Alix, C. Pérollier and S. Bayoudh, Anal. Chim. Acta, 2010, 672, 15–19 CrossRef CAS PubMed.
- J. L. Urraca, M. D. Marazuela and M. C. Moreno-Bondi, Anal. Bioanal. Chem., 2006, 385, 1155–1161 CrossRef CAS PubMed.
- R. Manova and R. Mladenova, Food Control, 2009, 20, 362–365 CrossRef CAS PubMed.
- D. Briones-Reyes, L. Gómez-Martinez and R. Cueva-Rolón, Food Chem., 2007, 100, 693–698 CrossRef CAS PubMed.
- F. Soleimany, S. Jinap, A. Faridah and A. Khatib, Food Control, 2012, 25, 647–653 CrossRef CAS PubMed.
- G. Fang, Ch. Fan, H. Liu, M. Pan, H. Zhu and Sh. Wang, RSC Adv., 2014, 4, 2764–2771 RSC.
- T. Li, B. B. Kim, W. B. Shim, J. Y. Byun, D. H. Chung, Y. B. Shin and M. G. Kim, Anal. Lett., 2014, 47, 453–464 CrossRef CAS.
- N. V. Beloglazova, E. S. Speranskaya, S. De Saeger, Z. Hens, S. Abé and I. Y. Goryacheva, Anal. Bioanal. Chem., 2012, 403, 3013–3024 CrossRef CAS PubMed.
- L. L. Wang, Sh. Yu and M. Yu, Spectrochim. Acta, Part A, 2012, 98, 337–342 CrossRef CAS PubMed.
- M. Hashemi, Z. Taherimaslak and S. Rashidi, Spectrochim. Acta, Part A, 2014, 128, 583–590 CrossRef CAS PubMed.
- H. Mine Antep and M. Merdivan, Anal. Methods, 2012, 4, 4129–4134 RSC.
- R. Romero-Gonzalez, J. L. M. Vidal, M. M. Aguilera-Luiz and A. Garrido Frenich, J. Agric. Food Chem., 2009, 57, 9385–9392 CrossRef CAS PubMed.
- A. Veršilovskis, B. Huybrecht, E. K. Tangni, L. Pussemier, S. De Saeger and A. Callebaut, Food Addit. Contam., 2011, 28, 1687–1693 Search PubMed.
- E. González-Peňas, C. Leache, M. Viscarret, A. Pérez de Obanos, C. Araguás and A. López de Cerain, J. Chromatogr. A, 2004, 1025, 163–168 CrossRef PubMed.
- M. Castegnarol, M. Tozlovanul, Ch. Wild, A. Molini, A. Sylla and A. Pfohl-Leszkowicz, Mol. Nutr. Food Res., 2006, 50, 480–487 Search PubMed.
- M. Hashemi, N. Jahanshahi and A. Habibi, Desalination, 2012, 288, 93–97 CrossRef CAS PubMed.
- M. Hashemi, S. M. Daryanavard and S. Abdolhosseini, Anal. Methods, 2013, 5, 6848–6854 RSC.
- A. Sarfraz-Yazdi and A. H. Amiri, Trends Anal. Chem., 2010, 29, 1–14 CrossRef PubMed.
- M. Rezaee, Y. Yamini and M. Faraji, J. Chromatogr. A, 2010, 1217, 2342–2357 CrossRef CAS PubMed.
- P. Hashemi, M. Naderlou, M. Safdarian and A. R. Ghiasvand, Anal. Chem. Lett., 2013, 3, 92–101 CrossRef CAS.
- A. Rahimi and P. Hashemi, J. Anal. Chem., 2014, 69, 352–356 CrossRef CAS.
- Zh. G. Shi and H. K. Lee, Anal. Chem., 2010, 82, 1540–1545 CrossRef CAS PubMed.
- Kh. S. Tay, N. A. Rahman and M. R. B. Abas, Anal. Methods, 2013, 5, 2933–2938 RSC.
- K. M. Giannoulis, D. L. Giokas, Q. Zhu, G. Z. Tsogas, A. G. Vlessidis and Q. Pan, Microchim. Acta, 2013, 180, 775–782 CrossRef CAS.
- M. Hashemi, Z. Taherimaslak and S. Rashidi, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 960, 200–208 CrossRef CAS PubMed.
- M. Hashemi and Z. Taherimaslak, RSC Adv., 2014, 4, 33497–33506 RSC.
- A. Beiraghi, K. Pourghazi and M. Amoli-Diva, Anal. Methods, 2014, 6, 1418–1428 RSC.
- JCPDS card 19-0629, Joint Committee on Powder Diffraction Standards, Swarth-more, 1989.
- L. Campone, A. L. Piccinelli, R. Celano and L. Rastrelli, J. Chromatogr. A, 2011, 1218, 7648–7654 CrossRef CAS PubMed.
- A. Mallick, P. Purkayastha and N. Chattopadhyay, J. Photochem. Photobiol., C, 2007, 8, 109–127 CrossRef CAS PubMed.
- M. Appell and W. B. Bosma, J. Lumin., 2011, 131, 2330–2334 CrossRef CAS PubMed.
- AOAC Official methods of analysis, Zearalenol and Zearalenone in Corn, AOAC International, Gaithersburg, MD, USA, Official Method 985.18, 17th edn, 2000.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra07684b |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.