
First reusable ligand-free palladium catalyzed C–P bond formation of aryl halides with trialkylphosphites in neat water†
Nasser Iranpoor*,
Habib Firouzabadi*,
Khashayar Rajabi Moghadam and
Somayeh Motavalli
Department of Chemistry, College of Sciences, Shiraz University, Shiraz 71454, Iran
First published on 7th October 2014
Abstract
A reusable ligand-free palladium catalyzed phosphonation of aryl iodides, bromides and chlorides with trialkylphosphites is described for the first time in neat water. The aryl phosphonates are obtained in good to excellent yields. The reaction can be also performed with Ni(II) with longer reaction time. The role of tetrabutylammonium bromide in this reaction as reducing agent for generation of Pd(0) at room temperature is also demonstrated. Pd(0)/TBAB was easily reused for three runs without decreasing the efficiency.
Introduction
Transition metal-catalyzed carbon–phosphorous bond formation plays a key role in the preparation of different types of phosphorous compounds such as phosphonates, phosphinates, phosphine oxides, phosphines, etc. Some of these compounds have been found wide biological activities or material science applications and some have been considered as versatile ligands in some catalytic reactions.1 In comparison with the phosphorus compounds containing the more reactive N–P, S–P or O–P linkages, organophosphonates are synthetically more important due to the more resistant of their C–P linkage to chemical hydrolysis, thermal decomposition and photolysis.2 Most of the methods for the synthesis of aryl phosphonates are usually based on the use of hydrogen dialkylphosphonates as the source of phosphorous compound. The first palladium-catalyzed phosphonation of aryl iodides or bromides was reported by Hirao et al. using this class of phosphorous compounds for C–P bond formatin.3a After this report, research efforts have focused on this reaction and remarkable progress have been achieved on the improvements of the classical Hirao reaction conditions such as; applying different transition metals1a,3,4,5 ligands and solvents3c,3d,3n,4c,6 alongside with use of techniques such as microwave irradiation.3h,7After successful application of dialkyl H-phosphonates as phosphorous source in phosphonation of aryl halides,3 the use of these phosphonating agents have been attended in C–P bond formation in the reaction with arylboronic acids,5b,8 terminal acetylenes,9 diaryliodoniumsalts3e,10 and sodium benzenesulfinates.11
In recent years, trialkylphosphites as convenient phosphorus source with higher boiling points and more chemical stability than dialkyl H-phosphonates have been found considerable synthetic applications in different organic transformations.12
The substitution of aryl halides with trialkylphosphites in the presence of stoichiometric amount of copper catalyst at 150–175 °C,13 the two step reaction of aniline with trialkylphosphites,14 the reaction of triethylphosphite with 2-haloanilides in the presence of NiX2 complexes that were conducted under the atmosphere of dry argon (Schlenk technique),15 doing reaction under microwave irradiation7 and using electrolysis methods16 are some examples for the synthesis of aryl phosphonates. In addition, recently Dhokale et al. reacted 2-(trimethylsilyl)phenyl trifluoromethanesulfonate as an unusual substrate instead of readily available aryl halides with triethylphosphite and cesium fluoride in acetonitrile under inert atmosphere at 20 °C to offer diethyl phenyl phosphonate after 20 h.17 The use of NiCl2 and PdCl2 with various ligands as catalyst have also been reported for preparation of diethylaryl phosphonates from aryl halides and trialkylphosphites but under high temperature at 140-–195 °C,18 inert atmosphere14,15 or microwave irradiation.7
The importance of green reactions in organic synthesis has encouraged scientists to use water as environmentally friendly reaction media. In this line, efforts have been focused on the aryl C–P bond formation in aqueous solutions. Reports on the C–P bond formation using aryl halides in neat water are rare in the literature.19,1a Tang's group reported the first example of the conversion of aryl iodides and bromides to aryl phosphine oxides using H-diphenylphosphine oxide in the presence of zinc powder and NiCl2·6H2O as the catalyst in water at 2011.19a On the other work, the reaction of halogenated benzoic acids were performed with H-diphenylphosphine oxide using Pd/C as the catalyst in water under microwave irradiation by Bokhoven et al.19b
Due to the importance of using water as the solvent for synthesis of phosphonates, Wu et al., subsequently developed the cross-coupling reaction of aryl halides by using diisopropyl H-phosphonate, and a palladacycle as catalyst in refluxing water-iso-propanol mixture at 2013.1a In general, dialkyl H-phosphonates are unstable in water and produce phosphorous acid and their corresponding alcohols. Regarding the reversibility of the decomposition reaction of diisopropyl H-phosphonate, to avoid this side reaction, Wu’s group added iso-propanol to the reaction mixture.1a As far as we know, there are no reports in the literature for the synthesis of phosphonates from aryl halides using trialkylphosphites in neat water.
In continuation of our recent works on the palladium catalyzed cross-coupling reactions,20 herein we report the first reusable ligand-free palladium catalyzed phosphorylation reaction of aryl halides with trialkylphosphites in neat water.
Results and discussion
Among the reported methods for phosphonation of aryl halides, the use of more stable trialkylphosphites with higher boiling points than their corresponding dialkyl H-phophonates seems to be a more practical method. In this work, for the first time, we report an efficient method for phosphonation of aryl iodides, and bromides as well as chlorides in the presence of trialkylphosphites in refluxing neat water as a green medium (Scheme 1).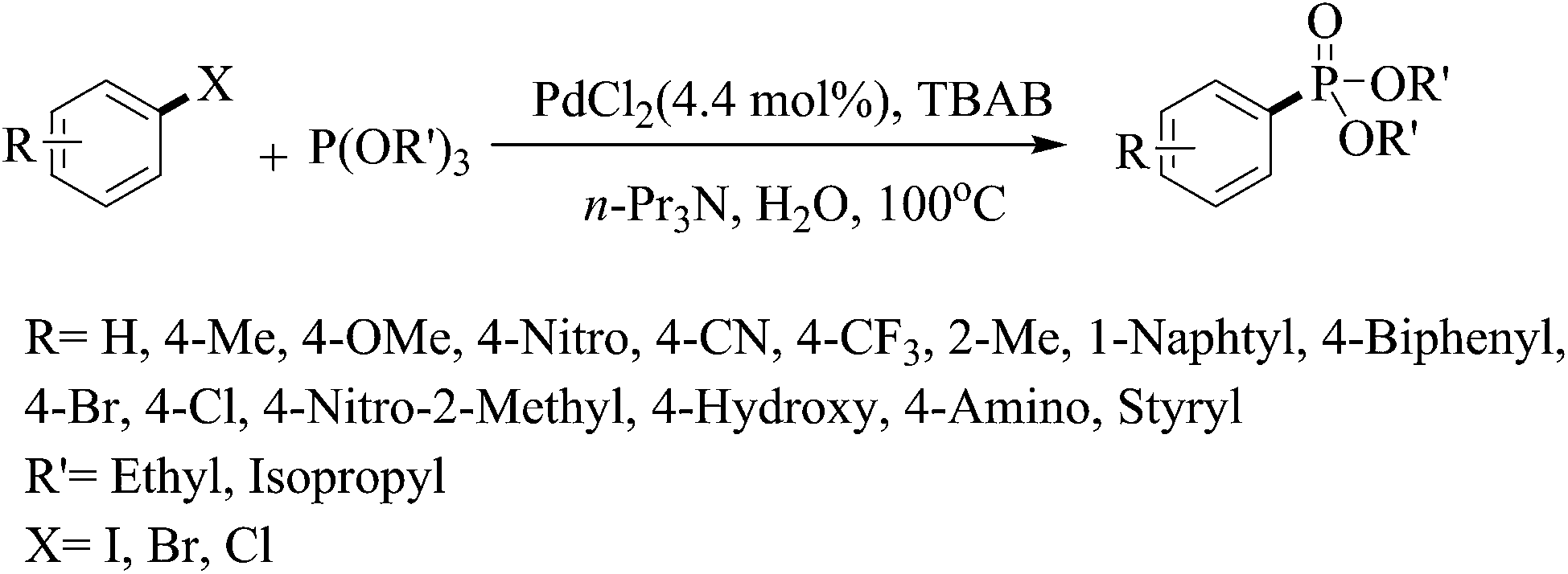 | ||
| Scheme 1 | ||
For optimization of the reaction conditions, we first performed the phosphonation of iodobenzene with triethylphosphite as a model reaction in the presence of catalytic amounts of PdCl2 and the effects of different parameters were studied. Several bases and solvents were screened for this reaction. Initially, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) was utilized as base and the reaction was carried out in the presence of tetrabutylammonium bromide (TBAB) in PEG 200 the solvent at 100 °C. The desired product was obtained in 90% yield after 12 h (Table 1, entry 1). Then DBU was replaced by bases such as 1,4-diazabicyclo[2.2.2]octane (DABCO), morpholine, Et3N, and KOAc. However, no improvement in the performance of the reaction was observed (Table 1, entries 2–5). Inorganic bases such as NaOH and K2CO3 were also found not to be suitable for this reaction in terms of reaction time and yield. (Table 1, entries 6 and 7). Using n-Pr3N as an organic base has pronounced effect and the reaction time was dramatically reduced from 12 h to 1.5 h in PEG200 and the yield was found to be excellent (Table 1, entry 8). The effect of other solvents such as DMF, DMSO, diglyme, EtOH, THF, H2O, and also solvent-free condition were then studied using n-Pr3N as the most suitable base (Table 1, entries 9–15). Among these solvents, H2O was found to be even more suitable than PEG200 and the reaction proceeded well in the shortest reaction time (Table 1, entry 14). Et3N was also examined as base in water, however, the yield of the product was decreased and instead, the yield of biphenyl was increased (Table 1, entry 23). When PdCl2 was replaced with NiCl2, monitoring of the reaction showed the formation of the desired product in excellent yield with some increase in the reaction time (Table 1, entry 16). In the absence of base, the starting material remained unchanged (Table 1, entry 22). After optimizing the solvent and the base, the required amount of TBAB was also optimized. The results of Table 1 show that decreasing the amount of TBAB from one equivalent to 0.25 extends the reaction time from 1 to 4 h (Table 1, entry 18). When TBAB was removed from the reaction mixture, the reaction was not occurred (Table 1, entry 19).
| Entry | Base | Solvent | Yieldb (%) | Time (h) |
|---|---|---|---|---|
| a Reaction conditions: 4-iodobenzene (0.5 mmol), triethylphosphite (2.0 mmol), base: (1.0 mmol), TBAB: (0.5 mmol), catalyst: (0.022 mmol), solvent (1.5 mL).b Isolated yield.c NiCl2 (0.022 mmol) as catalyst.d TBAB: (1.0 mmol).e TBAB: (0.25 mmol).f Without TBAB.g Base: (3.0 mmol).h Base (2.0 mmol).i Without base. | ||||
| 1 | DBU | PEG200 | 90 | 12 |
| 2 | DABCO | PEG200 | 84 | 12 |
| 3 | Morpholine | PEG200 | 70 | 24 |
| 4 | Et3N | PEG200 | 57 | 24 |
| 5 | KOAc | PEG200 | 35 | 24 |
| 6 | NaOH | PEG200 | 50 | 24 |
| 7 | K2CO3 | PEG200 | 66 | 24 |
| 8 | n-Pr3N | PEG200 | 90 | 1.5 |
| 9 | n-Pr3N | DMF | 70 | 24 |
| 10 | n-Pr3N | DMSO | 62 | 24 |
| 11 | n-Pr3N | Diglyme | 90 | 24 |
| 12 | n-Pr3N | Ethanol | 85 | 24 |
| 13 | n-Pr3N | THF | 45 | 2 |
| 14a | n-Pr3N | H2O | 98 | 1 |
| 15 | n-Pr3N | No-solvent | 76 | 2.5 |
| 16c | n-Pr3N | H2O | 90 | 5 |
| 17d | n-Pr3N | H2O | 96 | 1 |
| 18e | n-Pr3N | H2O | 95 | 4 |
| 19f | n-Pr3N | H2O | 0 | 24 |
| 20g | n-Pr3N | H2O | 97 | 1 |
| 21h | n-Pr3N | H2O | 96 | 1 |
| 22i | None | H2O | 0 | 24 |
| 23 | Et3N | H2O | 65 | 2 |
Having the optimized conditions; 0.5 mmol of aryl halide, 2.0 mmol of P(OR)3, 1.0 mmol of n-Pr3N, 0.022 mmol of PdCl2, and 0.5 mmol of TBAB in 1.5 mL of H2O at 100 °C (Table 1, entry 14), the phosphorylation reactions proceeded efficiently for iodobenzene and iodobenzenes bearing either an electron-withdrawing or an electron-donating group at the para-position (Table 2, entries 3a, 3b and 3d–f). The methodology was further extended to the coupling of sterically hindered aryl iodides such as 2-iodotoluene, 1-iodonaphthalene, and 4-nitro-2-iodotoluene (Table 2, entries 3g, 3h, and 3l). The applicability of this system was developed to aryl bromides and vinyl bromide. β-Bromostyrene and 1-bromonaphthalene reacted successfully to produce their corresponding phosphonates (Table 2, entries 3a′–e′, 3h′, 3i and 3o). In order to investigate the chemoselectivity of the reaction, 4-chlorobromobenzene and 4-bromoiodobenzene were examined under the optimized conditions and the results showed high selectivity as illustrated in Table 2, entries 3j and 3k. We also tried to use this method for the coupling of triethylphosphite with chlorobenzene. Although the cross-coupling reaction proceeded efficiently for chlorobenzenes bearing an electron-withdrawing group at the para-position (Table 2, entries 3d′′ and 3e′′), the reaction did not proceed with chlorobenzene, and chlorobenzenes bearing an electron-donating group even in a sealed tube. The reactions didn't performed even in the presence of PPh3 as ligand with increasing the amounts of PdCl2 from 5 mol% to 10 mol%. In entries 3m, 3m′, 3n and 3n′, the effect of hydroxy and amino groups in iodo and bromo derivatives of phenol and aniline were studied. The reactions proceeded well and their corresponding phosphonates were obtained in 79–90% yield.
|
|---|
| a Reaction conditions: aryl halide (0.5 mmol), trialkylphosphite (2.0 mmol), n-Pr3N (1.0 mmol), TBAB: (0.5 mmol), PdCl2 (4.4 mol%), solvent (1.5 mL). All yields are isolated yield. |
 |
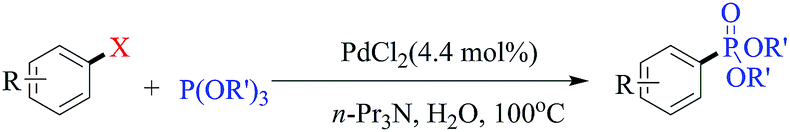
Instead of triethylphosphite, we also examined this method for the coupling of triisopropylphosphite with iodobenzene. The reactions were occurred efficiently and the corresponding products were obtained in high yields (Table 2, entries 3p–s).
The role of TBAB in this reaction is crucial. It is noteworthy that the presence of TBAB not only makes the catalytic system stable by preventing the formation of palladium black21 but also acts both as reducing agent for the generation of Pd(0) and also phase transfer catalyst for making the reaction mixture homogeneous.
In order to gain insight about the reducing property of TBAB, for the first time we probed the UV spectrum of the aqueous solution of Pd(II) in the presence of TBAB, n-Pr3N and also triethylphosphite separately. The UV spectra of these solutions are shown in Fig. 1. The white color of the aqueous solution of TBAB changed almost immediately to gray upon the addition of Pd(II) at room temperature. The UV spectrum showed the disappearance of the peak around 300 nm (blue curve) after 5 minutes, which confirms that Pd(II) has been reduced to the Pd(0) species. The formation of a peak at about 380 nm for Br2, demonstrates the efficiency of the generation of zero-valent palladium by TBAB through oxidation of bromide ion to Br2. In comparison, neither n-Pr3N, nor triethylphosphite reduced Pd(II) under similar condition (Fig. 1).
 | ||
| Fig. 1 UV spectra of PdCl2 in neat water with TBAB (blue), n-Pr3N (red) and P(OEt)3 (green) after 30 minutes at room temperature. | ||
The reduction of Pd(II) to Pd(0) in neat water at room temperature by TBAB is shown in Fig. 2. The isosbestic point at 358 nm shows the pretense of both Pd(II) species and bromine molecule in the solution.
 | ||
| Fig. 2 UV spectra of PdCl2 in neat water with TBAB at different time intervals at room temperature. | ||
This interesting property of TBAB to reduce Pd(II) to Pd(0) is in similar to the reducing properties of tetraalkylammonium carboxylates22a at 60 °C and tetrabutylammonium boronate22b in which instead of Br−, RCO2− and boronate anions have been responsible for electron transfer to Pd(II).
The recyclability of this system was also examined in the reaction of iodobenzene. For this purpose, after completion of the reaction and extraction of the product in ethyl acetate, iodobenzene, base and triethylphosphite were added to the aqueous layer containing Pd(0)/TBAB and the reaction mixture was worked-up after 1.5 h. Quantitative conversion to the corresponding phosphonate was observed for three runs. From the fourth run, loss of the activity of the catalytic system was observed (Fig. 3).
 | ||
| Fig. 3 Recyclability of Pd(0)/TBAB in the reaction of iodobenzene with triethylphosphite in water. | ||
The catalytic cycle for the phosphorylation reaction involves a series of transformations around the palladium catalyst. The proposed mechanism is as the following. According to the UV spectrum (Fig. 1), Pd(0) could well be generated by TBAB. Step 1 involves an oxidative addition in which Pd inserts into the aryl halide bond and the reaction continues to perform homogeneously in water. In step 2, triethylphosphite reacts with the palladium-halide bond and then by an Arbuzov type reaction, EtX is eliminated from adduct I which is generated from step 2 followed by the formation of Ar–Pd–phosphonate II. In step 4, the Pd(0) catalyst is regenerated by the reductive elimination of the palladium(II) compound and the corresponding aryl phosphonate is formed (Scheme 2).
 | ||
| Scheme 2 | ||
Experimental section
1H and 13C spectra were recorded on a Bruker Avance DPX-250 spectrometer and 31P NMR (on a Bruker UltraSheild-400 spectrometer) using tetramethylsilane (TMS) as internal standard in pure deuterated solvents. The reaction monitoring was carried out on silica gel analytical sheets or by GC analysis using a 1 m length column packed with DC-200 stationary phase. Column chromatography was carried out on a column of silica gel. The spectral data for the products are included in the ESI.†General procedure for phosphonation of aryl halides with trialkylphosphites using PdCl2 as catalyst in neat water
A mixture of PdCl2 (0.004 g, 0.022 mmol, 4.4 mol%), aryl halide (0.5 mmol), trialkylphosphite (2.0 mmol), n-Pr3N (1.0 mmol, 0.18 mL), TBAB (0.5 mmol, 0.166 g), in water (1.5 mL) was heated at 100 °C. After completion of the reaction monitored by GC or TLC analysis, the reaction mixture was cooled to room temperature. The organic compound was extracted with ethyl acetate (3.0 × 10 mL) from the aqueous layer and dried over anhydrous Na2SO4, filtered, and concentrated in vacuum. The crude organic mixture was then purified by silica gel column chromatography using petroleum ether–ethyl acetate 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as eluent to obtain the desired product (Table 1).
:1 as eluent to obtain the desired product (Table 1).
Conclusions
In conclusion we have reported the first reusable ligand-free Pd(0) catalytic method for the efficient phosphonation of aryl halides (X = Cl, Br, I) and also styryl bromide with different trialkylphosphites in neat water. The role of TBAB as reducing agent in water for the generation of Pd(0) was also demonstrated for the first time.Acknowledgements
We are grateful to the research council of Shiraz University and a grant from Iran National Elite Foundation for the support of this work.Notes and references
- (a) K. Xu, F. Yang, G. Zhang and Y. Wu, Green Chem., 2013, 15, 1055 RSC; (b) D. S. Glueck, Top. Organomet. Chem., 2010, 31, 65 CrossRef CAS; (c) C. S. Demmer, N. K. Larsen and L. Bunch, Chem. Rev., 2011, 111, 7981 CrossRef CAS PubMed.
- N. G. Ternan, J. W. McGrath, G. McMullan and J. P. Quinn, World J. Microbiol. Biotechnol., 1998, 14, 635 CrossRef CAS.
- For palladium-catalyzed sp2 hybridized C–P bond formation, see: (a) T. Hirao, T. Masunaga, Y. Ohshiro and T. Agawa, Tetrahedron Lett., 1980, 21, 3595 CrossRef CAS; (b) T. Hirao, T. Masunaga, N. Yamada, Y. Ohshiro and T. Agawa, Bull. Chem. Soc. Jpn., 1982, 55, 909 CrossRef CAS; (c) D. Gelman, L. Jiang and S. L. Buchwald, Org. Lett., 2003, 5, 2315 CrossRef CAS PubMed; (d) L. J. Gooβen and M. K. Dezfuli, Synlett, 2005, 445 Search PubMed; (e) S. Thielges, P. Bisseret and Eustache, Org. Lett., 2005, 7, 681 CrossRef CAS PubMed; (f) K. Bravo-Altamirano, Z. H. Huang and J. L. Montchamp, Tetrahedron, 2005, 61, 6315 CrossRef CAS; (g) M. Kalek and J. Stawinski, Organometallics, 2007, 26, 5840 CrossRef CAS; (h) M. Kalek, A. Ziadi and J. Stawinski, Org. Lett., 2008, 10, 4637 CrossRef CAS PubMed; (i) M. Kalek and J. Stawinski, Organometallics, 2008, 27, 5876 CrossRef CAS; (j) Y. Belabassi, S. Alzghari and J. L. Montchamp, J. Organomet. Chem., 2008, 693, 3171 CrossRef CAS PubMed; (k) A. Bessmertnykh, C. M. Douaihy, S. Muniappan and R. Guilard, Synthesis, 2008, 10, 1575 Search PubMed; (l) L. Coudray and J. L. Montchamp, Eur. J. Org. Chem., 2008, 4101 CrossRef CAS PubMed; (m) M. Kalek, M. Jezowska and J. Stawinski, Adv. Synth. Catal., 2009, 351, 3207 CrossRef CAS; (n) A. Bessmertnykh, C. M. Douaihy and R. Guilard, Chem. Lett., 2009, 38, 738 CrossRef CAS; (o) Y. Luo and J. Wu, Organometallics, 2009, 28, 6823 CrossRef CAS; (p) D. Hockova, M. Dracinsky and A. Holy, Eur. J. Org. Chem., 2010, 2885 CrossRef CAS.
- For copper-catalyzed C–P bond formation, see: (a) T. Ogawa, N. Usuki and N. Ono, J. Chem. Soc., Perkin Trans. 1, 1998, 2953 RSC; (b) H. H. Rao, Y. Jin, H. Fu, Y. Y. Jiang and Y. F. Zhao, Chem.–Eur. J., 2006, 12, 3636 CrossRef CAS PubMed; (c) C. Huang, X. Tang, Y. Y. Jiang and Y. F. Zhao, J. Org. Chem., 2006, 71, 5020 CrossRef CAS PubMed; (d) B. Xiong, M. Li, Y. Liu, Y. Zhou, C. Zhao, M. Goto, S. F. Yin and L. Han, Adv. Synth. Catal., 2014, 356, 781 CrossRef CAS.
- (a) Y. L. Zhao, G. J. Wu, Y. Li, L. X. Gao and F. S. Han, Chem.–Eur. J., 2012, 18, 9622 CrossRef CAS PubMed; (b) W. Chen, T. Fu, Y. Gao, G. Hu, Z. Peng, H. Qiao and Y. Zhao, Org. Lett., 2013, 15, 5362 CrossRef PubMed.
- H. Hu, Y. Wu, K. Xu and F. Yang, Eur. J. Org. Chem., 2013, 2, 319 Search PubMed.
- (a) D. Villemen, A. Elbilali, F. Simeon, P. A. Jaffres, G. Maheut, M. Mosaddak and A. Hakiki, J. Chem. Res., Synop., 2003, 436 CrossRef; (b) E. Jablonkai and G. Keglevich, Tetrahedron Lett., 2013, 54, 4185 CrossRef CAS; (c) G. Keglevich, A. Gruen, A. Boelcskei, A. Gruen, L. Drahos, M. Kraszni and G. T. Balogh, Heteroat. Chem., 2012, 23, 574 CrossRef CAS; (d) A. F. Gurley and J. J. Kiddle, Phosphorus, Sulfur Silicon Relat. Elem., 2000, 160, 195 CrossRef.
- (a) R. Zhuang, J. Xu, Z. Cai, G. Tang, M. Fang and Y. Zhao, Org. Lett., 2011, 13, 2110 CrossRef CAS PubMed; (b) M. Andaloussi, M. Larhed, J. Lindh, J. Saevmarker and P. J. R. Sjoeberg, Chem.–Eur. J., 2009, 15, 13069 CrossRef CAS PubMed.
- Y. Gao, G. Wang, L. Chen, P. Xu, Y. Zhao, Y. Zhou and L.-B. Han, J. Am. Chem. Soc., 2009, 131, 7956 CrossRef CAS PubMed.
- (a) C. Zhou, Synth. Commun., 2001, 31, 3289 CrossRef; (b) C. Liu, Synthesis, 1993, 4, 373 CrossRef.
- T. Miao and L. Wang, Adv. Synth. Catal., 2014, 356, 967 CrossRef CAS.
- (a) G. R. Qu, R. Xia, X. N. Yang, J. G. Li, D. C. Wang and H. M. Guo, J. Org. Chem., 2008, 73, 2416 CrossRef CAS PubMed; (b) R. J. Barney, R. M. Richardson and D. F. Wiemer, J. Org. Chem., 2011, 76, 2875 CrossRef CAS PubMed; (c) B. Das, K. Damodar and N. Bhunia, J. Org. Chem., 2009, 74, 5607 CrossRef CAS PubMed; (d) G. G. Rajeshwaran, M. Nandakumar, R. Sureshbabu and A. K. Mohanakrishnan, Org. Lett., 2011, 13, 1270 CrossRef CAS PubMed.
- (a) G. Axelrad, S. Laosooksathit and R. Engel, J. Org. Chem., 1981, 46, 5200 CrossRef CAS; (b) N. Hall and R. J. Price, J. Chem. Soc., Perkin Trans. 1, 1979, 2634 RSC.
- R. Berrino, S. Cacchi, G. Fabrizi, A. Goggiamani and P. Stabile, Org. Biomol. Chem., 2010, 8, 4518 CAS.
- J. Heinicke, N. Gupta, A. Surana, N. Peulecke, B. Witt, K. Steihauser, R. R. Bansal and P. G. Yones, Tetrahedron, 2001, 57, 9963 CrossRef CAS.
- (a) M. Rafiee, S. M. Shoaei and L. Khalafi, Electrochim. Acta, 2012, 80, 56 CrossRef CAS; (b) Y. M. Kargin, E. V. Nikitin, O. V. Parakin, G. V. Romanov and A. N. Pudovik, Phosphorus, Sulfur Silicon Relat. Elem., 1980, 8, 55 CrossRef CAS.
- R. A. Dhokale and S. B. Mhaske, Org. Lett., 2013, 15, 2218 CrossRef CAS PubMed.
- (a) V. V. Sentemov, E. A. Krasil'nikova and I. V. Berdnik, J. Gen. Chem. U. S. S. R., 1989, 59, 2406 Search PubMed; (b) C. Yuan and H. Feng, Synthesis, 1990, 2, 140 CrossRef; (c) T. M. Balthazor and R. C. Grabiak, J. Org. Chem., 1980, 5425 CrossRef CAS; (d) C. Shen, G. Yang, L. Zhang and W. Zhang, Tetrahedron Lett., 2011, 52, 5032 CrossRef PubMed.
- For sp2 hybridized C–P bond formation in water, see: (a) X. H. Zhang, H. Z. Liu, X. M. Hu, G. Tang, J. Zhu and Y. F. Zhao, Org. Lett., 2011, 13, 3478 CrossRef CAS PubMed; (b) S. M. Rmmelt, M. Ranochiari and J. A. Bokhoven, Org. Lett., 2012, 14, 2188 CrossRef PubMed.
- (a) N. Iranpoor, H. Firouzabadi and S. Motevalli, J. Mol. Catal. A: Chem., 2012, 355, 69 CrossRef CAS; (b) N. Iranpoor, H. Firouzabadi, S. Motevalli and M. Talebi, J. Organomet. Chem., 2012, 708, 118 CrossRef.
- (a) J. H. Li, W. J. Liu and Y. X. Xie, J. Org. Chem., 2005, 70, 5409 CrossRef CAS PubMed; (b) M. Beller, H. Fischer, K. Kiihlein, C. P. Reisinger and W. A. Herrmarm, J. Organomet. Chem., 1996, 520, 257 CrossRef CAS; (c) W. J. Liu, Y. X. Xie, Y. Liang and J. H. Li, Synthesis, 2006, 5, 860 CrossRef.
- (a) M. T. Reetz and E. Westermann, Angew. Chem., Int. Ed., 2000, 39, 165 CrossRef CAS; (b) H. Bonnemann, W. Brijoux, R. Brinkmann and E. T. Denjus, Angew. Chem., 1991, 103, 1344 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra07680j |
| This journal is © The Royal Society of Chemistry 2014 |