DOI:
10.1039/C4RA07614A
(Paper)
RSC Adv., 2014,
4, 52324-52331
Developing thioxanthone based visible photoinitiators for radical polymerization
Received
25th July 2014
, Accepted 13th October 2014
First published on 14th October 2014
Abstract
Three new amino-thioxanthone photoinitiators (PIs) with different substituents on nitrogen, including 2-allyl(methyl)amino-9H-thioxanthen-9-one (PI-1), 2-benzyl(methyl)amino-9H-thioxanthen-9-one (PI-2) and 2-butyl(methyl)amino-9H-thioxanthen-9-one (PI-3) were synthesized and characterized. They showed high molar extinction coefficients and very broad absorption range in the visible region. Among them, PI-2 showed the best visible light photoinitiating properties evidenced by the fact that it successfully initiated polymerization of 1,6-hexanedioldiacrylate (HDDA) under xenon light exposure (28 mW cm−2) both in the presence and absence of N,N-dimethylaniline (DMA) for 0.5 h with 92% and 52% conversion, respectively. For TMPTA, the conversion of PI-2 and PI-2/N-methyldiethanolamine (MDEA) systems is 32% and 62% under xenon lamp exposure for 10 min. The photoinitiation mechanisms were analysed through EPR, fluorescence spectra and visible light photolysis experiments.
Introduction
Photoinitiated polymerizations have been the subject of increasing interest because of their widespread applications ranging from imaging and optic technologies to medicine, microelectronics, nanotechnology, and material elaboration areas.1–4 As one of the most important parts in photopolymerization system, photoinitiator absorbs UV or visible light radiations and produces primary active species which trigger the polymerization or crosslinking reaction.1,2,5 According to their sensitive wavelength, radical photoinitiators can be divided into UV photoinitiators and visible photoinitiators. The UV photoinitiators have been widely used in industry fields such as coatings, inks, and photoresistors, etc. Recently, the visible photoinitiators have attracted much more attention not only because that visible light is cheap, safety and possesses high penetration ability in the presence of ultraviolet absorbing monomers, pigments, and substrates but also visible photopolymerization has many targeted applications such as printing plates,6 integrated circuits,7 dental filling materials,8,9 photoresists,10 laser-induced 3D curing,11 holographic recordings,11 and nanoscale micromechanics.12 Bis(cyclopentadienyl)titanium(IV) dichloride13 and iridium complexes14 have been used as visible photoinitiators. Besides, push-pull Michler's ketone,15 malonate and malononitrile16 based dyes have been investigated as photosensitizers in the presence of free radical sources.
Among a lot of photoinitiators, thioxanthone derivatives in conjunction with tertiary amines are one of the most investigated chromophores which mainly absorb in the UV part of the electromagnetic spectrum. As a type II photoinitiator, thioxanthone derivatives suffer from the distinct disadvantage as the requirement of hydrogen donors such as tertiary amines, thiols, alcohols and ethers to yield the initiating radicals.17–19 Although alkylamines are very efficient hydrogen donors, the high usage of such highly volatile and odorous compounds brings disadvantages to type II systems.
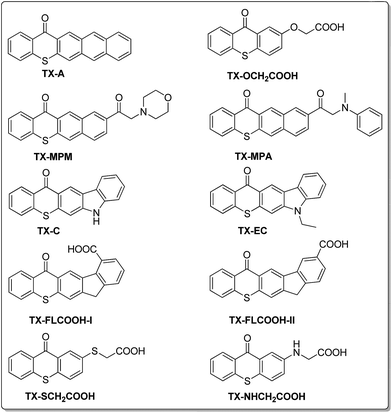
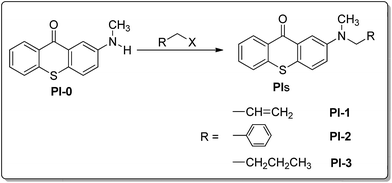
Several new thioxanthone derivatives as photoinitiators for free radicals polymerization have been reported to avoid problems associated with the amine hydrogen donors. Poly(ethylene glycol)-thioxanthone20 and poly(ethylene imine)-thioxanthone21 photoinitiator have been successfully used as polymeric hydrogen donors to replace low-molecular-weight amines. TX-A22,23 (Chart 1) and thioxanthone-diphenyl anthracene24 initiates the polymerization of acrylate and styrene monomers in the presence of air. Intra- or inter-molecular hydrogen abstraction reaction followed by decarboxylation process gave a convenient method to form initiating radicals, such as TX-OCH2COOH,25–27 TX-SCH2COOH,25–27 TX-FLCOOH28,29 and TX-NHCH2COOH30 (Chart 1). Meanwhile, chemically incorporating the hydrogen donating sites into the TX structure such as TX-MPM,31 TX-MPA,32 TX-C33,34 and TX-EC35 (Chart 1) provides an efficient strategy to form a one-component photoinitiator system. Interestingly, these photoinitiators do not require an additional co-initiator, that is, a separate molecular hydrogen donor, since the hydrogen donor is incorporated into the photoinitiator structure. Following this idea, the introduction of allyl-, benzyl- or butyl-group in 2-methylamino-9H-thioxanthen-9-one (PI-0) should not only provide another α-amino radical as hydrogen donor, but also bring about the redshift to visible region. Herein, a series of TX-based one-component radical polymerization visible photoinitiators PIs, 2-allyl(methyl)amino-9H-thioxanthen-9-one (PI-1), 2-benzyl(methyl)amino-9H-thioxanthen-9-one (PI-2) and 2-butyl(methyl)amino-9H-thioxanthen-9-one (PI-3) (Scheme 1) have been prepared and their photophysical and photochemical properties have been investigated.
 |
| | Chart 1 | |
 |
| | Scheme 1 | |
Experimental
Materials
2-Methylamino-9H-thioxanthen-9-one (PI-0) and 1,6-hexanedioldiacrylate (HDDA) were prepared following the literature method.36,37 All reagents and solvents were obtained from Aladdin and used as received without further purification. 1H (300 MHz) and 13C (75 MHz) NMR spectra were determined at room temperature on a VARIAN Mercury 300 spectrometer of the Spectropole. Mass spectrometry was carried out using a Varian 320-MS triple quadrupole mass spectrometer operated in the electron ionization (EI) mode.
Synthesis of 2-allyl(methyl)amino-9H-thioxanthen-9-one (PI-1)
To a mixture of PI-0 (2.41 g, 10 mmol) and 3-chloroprop-1-ene (1.14 g, 15 mmol) in toluene (20 mL), 20 mL of a 50% KOH solution and tetrabutylammonium bromide (0.17 g) was added. The mixture was heated with stirring at 90 °C until the absence of PI-0 (TLC test). Generally, this required at least 6 h. The organic layer was separated and washed with water (30 mL × 3). The organic phase was dried over anhydrous MgSO4. The residue was washed with petroleum ether and dried under vacuum after the solvent was removed. 2.59 g of a yellow solid was obtained in 92% yield, mp 119–120 °C. 1H NMR (CDCl3) δ (ppm): 3.08 (s, 3H, N–CH3), 4.04 (d, J = 4.8 Hz, 2H, N–CH2), 5.15–5.20 (m, 2H, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.82–5.91 (m, 1H, –CHCH2), 7.11 (dd, J1 = 3.0 Hz, J2 = 8.4 Hz, 1H, Ar H), 7.43–7.46 (m, 2H, Ar H), 7.56–7.58 (m, 2H, Ar H), 7.88 (d, J = 3.0 Hz, 1H, Ar H), 8.63 (d, J = 8.4 Hz, 1H, Ar H). 13C NMR (CDCl3) δ (ppm): 179.8, 147.8, 137.6, 132.8, 131.4, 129.6, 128.5, 126.6, 125.8, 125.3, 123.7, 118.5, 116.3, 110.2, 54.8, 38.0. MS, m/z (EI) 281.2 ([M + H]+, 100%).
CH2), 5.82–5.91 (m, 1H, –CHCH2), 7.11 (dd, J1 = 3.0 Hz, J2 = 8.4 Hz, 1H, Ar H), 7.43–7.46 (m, 2H, Ar H), 7.56–7.58 (m, 2H, Ar H), 7.88 (d, J = 3.0 Hz, 1H, Ar H), 8.63 (d, J = 8.4 Hz, 1H, Ar H). 13C NMR (CDCl3) δ (ppm): 179.8, 147.8, 137.6, 132.8, 131.4, 129.6, 128.5, 126.6, 125.8, 125.3, 123.7, 118.5, 116.3, 110.2, 54.8, 38.0. MS, m/z (EI) 281.2 ([M + H]+, 100%).
Synthesis of 2-benzyl(methyl)amino-9H-thioxanthen-9-one (PI-2)
A mixture of PI-0 (2.41 g, 10 mmol), benzyl chloride (1.32 g, 10.5 mmol), K2CO3 (2.76 g, 20 mmol) and 20 mL of DMF was refluxed until the absence of PI-0 (TLC test). Then 20 mL of water was added and extracted with ethyl acetate (40 mL × 4). The organic layer was collected, and dried over anhydrous MgSO4. The residue was washed with petroleum ether and dried under vacuum after the solvent was removed. 2.98 g of a yellow solid was obtained in 90% yield, mp 152–153 °C. 1H NMR (CDCl3) δ (ppm): 3.15 (s, 3H, N–CH3), 4.70 (s, 2H, N–CH2), 7.21–7.24 (m, 3H, Ar H), 7.30–7.35 (m, 3H, Ar H), 7.53 (t, J = 7.2 Hz, 1H, Ar H), 7.62–7.78 (m, 4H, Ar H), 8.43 (d, J = 7.2 Hz, 1H, Ar H). 13C NMR (CDCl3) δ (ppm): 180.0, 148.2, 138.0, 137.6, 131.5, 127.7, 128.5, 127.0, 126.7, 126.5, 125.9, 125.5, 124.1, 118.7, 110.3, 56.2, 38.8. MS, m/z (EI) 297.3 ([M + H]+, 95%).
Synthesis of 2-butyl(methyl)amino-9H-thioxanthen-9-one (PI-3)
It was synthesized following the method of PI-1 and purified by SiO2 column chromatography. A yellow solid was obtained in 80% yield, mp 47–49 °C. 1H NMR (CDCl3) δ (ppm): 0.96 (t, J = 7.2 Hz, 3H, –CH2CH3), 1.31–1.44 (m, 2H, –CH2CH2CH3), 1.55–1.65 (m, 2H, N–CH2CH2CH2–), 3.05 (s, 3H, N–CH3), 3.43 (t, J = 7.2 Hz, 2H, N–CH2CH2–), 7.11 (dd, J1 = 3.0 Hz, J2 = 9.0 Hz, 1H, Ar H), 7.43–7.46 (m, 2H, Ar H), 7.57 (d, J = 3.6 Hz, 2H, Ar H), 7.85 (d, J = 3.0 Hz, 1H, Ar H), 8.63 (d, J = 9.0 Hz, 1H, Ar H). 13C NMR (CDCl3) δ (ppm): 179.8, 147.7, 137.7, 131.3, 129.6, 128.4, 126.5, 125.8, 125.3, 123.0, 118.2, 109.8, 52.1, 38.2, 28.6, 20.1, 13.9. MS, m/z (EI) 330.6 ([M + H]+, 100%).
Light source
The light source was assembled from xenon lamp (laite optics, XD 300, cold light source) with a filter (λ > 400 nm). The light intensity was determined using a SRC-1000-TC-QZ-N reference monocrystalline silicon cell system (Oriel, USA), which was calibrated by National Renewable Energy Laboratory, A2LA accreditation certificate 2236.01. Two kinds of different irradiation intensity were used: 28 and 57 mW cm−2.
Photopolymerization experiments
HDDA and TMPTA were used as low viscosity monomers. N,N-Dimethylaniline (DMA) and N-methyldiethanolamine (MDEA) were used as hydrogen donors. The film polymerization experiments were carried out in laminated conditions. The photosensitive formulations were deposited on a KBr pellet in laminate for irradiation with xenon lamp (I = 28 mW cm−2). Monomers, PIs and hydrogen donors in THF were acted as photopolymerization system. The evolution of the double-bond content was continuously monitored by real time FT-IR spectroscopy (Nicolet 60-SXB) at 1610–1650 cm−1. The degree of conversion is calculated from the equation:
where A0 and At represent the area of the IR absorption peak at 1610–1650 cm−1 of the sample before and after exposure during time t.
EPR spin trapping (EPR-ST) experiments
EPR spin-trapping experiments were carried out using a Bruker X-band A200. The radicals were produced at room temperature under a xenon lamp irradiation (28 mW cm−2) and trapped by phenyl-N-tert-butylnitrone (PBN). EPR spectra were recorded at 298 K on EPR spectrometer operated at 9.399 GHz. Typical spectrometer parameters are shown as follows, scan range: 100 G; center field set: 3354.8 G; time constant: 163.84 ms; scan time: 40.96 s; modulation amplitude: 1.0 G; modulation frequency: 100 kHz; receiver gain: 1.00 × 104; microwave power: 17.26 mW. The EPR spectrum simulations were carried out with the Biomolecular EPR Spectroscopy software. The aN and aH stand for the hyperfine coupling constants in the PBN radical adducts for the nitrogen and the hydrogen, respectively.
Visible light photolysis experiments
The photodecomposition of photoinitiators was followed by using the changes in absorbance at the wavelength of maximum absorption in visible region. The absorption spectra of photoinitiators were measured with a UV-visible spectrophotometer (Agilent 8453) in the THF solution under xenon lam exposure (57 mW cm−2) at room temperature in the presence of DMA or not.
Fluorescence experiments
The fluorescence properties of photoinitiators (PIs) were determined in THF using a spectrometer (RF-5301PC) and the quenching constants were obtained from a Stern–Volmer treatment1,2 I0/I = 1 + Ksv[Q], where I0 and I stand for the fluorescence intensity of the photoinitiator in the presence and absence of the quencher Q, respectively and Ksv is the Stern–Volmer quenching constant in M−1.
Results and discussion
Synthesis and characterization
Photoinitiators (PIs) were prepared by a convenient substituent reaction of PI-0, which was easily obtained following the literature procedure. The structures of photoinitiators (PIs) were confirmed by 1H NMR, 13C NMR and MS. Three photoinitiators (PIs) show high solubility in common polar and nonpolar solvents, such as THF, ethanol, acetonitrile, acetone, ethyl acetate and petroleum ether, demonstrating that they have excellent compatibility with other components of photopolymerization system.
Absorption and fluorescence
UV-vis absorption spectra of photoinitiators in THF have been shown in Fig. 1. The maximum absorption wavelength (λmax) and the molar extinction coefficients (ε) in visible region have been summarized in Table 1. The maximum absorption wavelength of PI-0 was 434 nm (ε = 4200 M−1 cm−1). As can be seen from Fig. 1 and Table 1, the introduction of allyl-, benzyl- and butyl-groups in PI-0 not only kept the high molar extinction coefficients (ε), but also brought about a redshift for PIs. Both PI-1 and PI-2 have strong absorption at 438 nm, and PI-3 has a λmax at 444 nm. All of them have broad absorptions evidenced by the fact that their absorption edges reach to 500 nm. Broad absorptions and high molar extinction coefficients (ε > 4000) are favorable for absorbing visible light and producing radicals to initiate radical polymerization.
 |
| | Fig. 1 UV-vis absorption spectra of PI-0 and PIs in THF solution. | |
Table 1 Photophysical data of PI-0 and PIsa
| Photoinitiators |
λmax (nm) |
ε (M−1 cm−1) |
λex (nm) |
λem (nm) |
| λmax is the maximum absorption wavelength in visible region. ε is the molar extinction coefficiency at λmax. λex is the excitation wavelength. λem is the emission wavelength. |
| PI-0 |
434 |
4200 |
442 |
517 |
| PI-1 |
438 |
4400 |
448 |
517 |
| PI-2 |
438 |
4800 |
448 |
512 |
| PI-3 |
444 |
4100 |
454 |
526 |
Fluorescence spectra of the photoinitiators may also provide information on the nature of the excited states involved. As can be seen from Table 1 and the Fig. 2, excitation and emission fluorescence spectra showed a nearly mirror-image relation between absorption and emission, indicating its dominant excited (singlet) state in the photoinitiator.
 |
| | Fig. 2 Fluorescence excitation and emission spectra of PIs in THF solution [8 × 10−6 mol L−1]. | |
The fluorescence quenching study of PIs was performed by the addition of DMA at different concentrations at room temperature. The changes in the fluorescence emissions were recorded at an excitation wavelength of 450 nm. The Stern–Volmer representation of this quenching process in THF has been shown in Fig. 3. The Stern–Volmer quenching constant, Ksv, which was correlated with the energetics of electron transfer between the singlet excited state of PIs and the quencher DMA38 and could be obtained from the slope, is 1.57 M−1 for PI-1, 2.09 M−1 for PI-2, and 0.29 M−1 for PI-3, respectively. Decrease of the fluorescence intensity is due to the formation of the nonfluorescent complex in the steady state. It is close to the rate constant of the diffusion-controlled reaction rate.39,40 In PIs and PIs/DMA systems, the rate of electron transfer and proton transfer may be lower in the order: PI-2, PI-1 and PI-3.
 |
| | Fig. 3 Stern–Volmer plot of the quenching of PIs [8 × 10−6 mol L−1] by DMA in THF. | |
Visible light photolysis
As demonstrated for previously reported highly conjugated TX derivatives,28,29,35,41 PIs are expected to undergo an irreversible photolysis in the presence and absence of hydrogen donors. This was further confirmed for PIs by the spectral changes to the absorption spectral on visible light irradiation. In Fig. 4, a bleaching of PIs and PIs/DMA solution is found upon the irradiation of xenon lamp. In Fig. 4(a), (c) and (e), the absorbance at 450 nm almost does not change at the beginning. The possible reason42–44 is that during irradiation PIs absorbs visible light due to the n–π* transition of the ketone carbonyl to produce an excited singlet state which passed into an excited triplet state (Scheme 2A) and interacts with hydrogen donors as itself to generate amine radical cations and oxygen radical anions (Scheme 2B). In the presence of air/O2, C and D (Scheme 2) may occur and back radical transformation may lead the photoinitiator to keep its structure.45–47 As yet, there is no positive evidence has been obtained for the formation of ground-state complexes among this system. And therefore the extent of participation of such species cannot be ascertained.
 |
| | Fig. 4 UV-vis spectral changes during irradiation of PIs [2 × 10−4 mol L−1] and PIs/DMA [2 × 10−4 mol L−1] in THF under xenon lamp exposure. | |
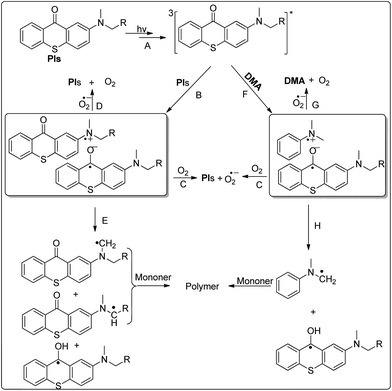 |
| | Scheme 2 Photoinitiated free radical polymerization mechanism by using PIs with and without a coinitiator. | |
Upon xenon lamp irradiation, the absorbance of PIs at 450 nm is decreased. It is ascribed to the transformation of amine radical cations and oxygen radical anions to a new ketyl radical (Scheme 2E) which is readily to undergo coupling reacting e.g. dimerization to give pinacols and combination with α-aminoalkyl radical. The conjugated system is damaged by the generation of new compounds. In Fig. 4(b), (d) and (f), the photolysis rate of PIs is more evident. The photoexcited PIs and DMA interact to form an exciplex (Scheme 2F). In the presence of air/O2, C and G (Scheme 2) may occur. The formation of DMA-derived, carbon-centered radical and ketyl radical (Scheme 2H) accelerates the degradation rate of the type II photoinitiators PIs. PIs were found to be an efficient initiator for visible photopolymerization in the presence and absence of a hydrogen donor. This is in accordance with the photo-bleaching results.
EPR spin trapping (EPR-ST)
From EPR-ST experiments, the formation of α-aminoalkyl radicals upon irradiation is clearly evidenced for PIs (Fig. 5). The hyperfine splitting (hfs) constants of the PBN adducts for PI-1 are aN = 14.2 G, aH = 2.5 G; for PI-2 are aN = 14.2 G, aH = 2.4 G; and for PI-3 are aN = 14.2 G, aH = 2.5 G. The measured aH values correspond to those reported for carbon-centered structures.16,48–50 The oxidized PBN is also observed with aN = 7.95 G, aH = 0.5 G; another PBN adduct is also observed and the hfs constants for PI-1 are aN = 14.2 G, aH = 5.2 G; for PI-2 are aN = 14.2 G, aH = 5.2 G; and for PI-3 are aN = 14.0 G, aH = 5.2 G. The EPR data further supports the formation of α-aminoalkyl radicals (Scheme 2E).
 |
| | Fig. 5 EPR spin trapping experiments of PIs (using PBN in tert-butyl-benzene under argon) upon xenon lamp irradiation (a) experimental and (b) simulated EPR spectra. | |
Mechanisms
On the basis of the above studies, the photochemical mechanism of the radical formation in PIs is illustrated in Scheme 2. It appears that the radicals are likely generated from the electron transfer followed by the proton transfer between the triplet PIs and the ground state PIs. The photochemical reaction takes into account the formation of an exciplex. It deactivates through back electron transfer without formation of any radical or leads to the formation of a ketyl radical and an initiating aminoalkyl radical. Ketyl radical typically does not initiate polymerization.51 The aminoalkyl radical initiates monomer radical polymerization in the absence of hydrogen donors. When DMA was added, the ground-state DMA and the photoexcited PIs interact to form an exciplex. A principal decay path for the exciplex is intermolecular hydrogen abstraction to form ketyl radical and amine-derived, carbon-centered radical. The radical is an efficient free-radical initiator which accelerates the rate of polymerization.
Photopolymerization
PIs were used as the photoinitiator for the polymerization of HDDA in laminate in the presence and absence of hydrogen donor under the xenon lamp exposure. Fig. 6(a) showed the visible photopolymerization of PIs/HDDA in the absence of hydrogen donor. The conversion increases continuously as the exposure time is varied in the range 0–0.5 h. At 0.5 h, the conversion of HDDA is 41%, 52% and 26%, respectively. In Fig. 6(b), the efficiency of the initiator process increases due to the formation of aminoalkyl radical in the presence of DMA. At 0.5 h, the conversion of HDDA is 77%, 92% and 67%, respectively. PIs/DMA behaves as type II photoinitiator system. The high concentration of DMA supplies high quantity hydrogen donors, which makes the electron/proton transformation faster than PIs photoinitiator system. The addition of a co-initiator (DMA) improves the performance of PIs. It is in line with the fluorescence quenching experiment. This is expected as hydrogen abstraction in this case that would lead to the formation of more stable benzyl radical and allyl radical.52 On the other hand, this can be partly ascribed to absorption of the photoinitiator. Overall, among all of the three, PI-2 has the best photoinitiating ability which contributes to introduce of benzyl amine.
 |
| | Fig. 6 Photopolymerization profiles of HDDA in laminate upon a xenon lamp exposure in the presence of (a) PI-1 (1%, w/w) (1), PI-2 (1%, w/w) (2), PI-3 (1%, w/w) (3); (b) PI-1/DMA (1%/5%, w/w) (1), PI-2/DMA (1%/5%, w/w) (2), PI-3/DMA (1%/5%, w/w) (3). | |
Since many literature15,16,48,53–55 results were obtained in TMPTA/MDEA couples system, for comparison, the ability of PI-2 for the initiation of the TMPTA photopolymerization was also tested and the results were illustrated in Fig. 7. In the absence of hydrogen donors, the conversion of double bond is 32% after 10 min under the xenon lamp exposure. Upon addition of DMA, the polymerization of TMPTA is enhanced with a conversion of 50%. For comparison, the MDEA was used as hydrogen donor. Interestingly, the PI-2/MDEA couples lead to a good polymerization profile: the final conversion reaches 62%. Jacques Lalevée group has reported anthraquinone derivatives55 and naphthalimide based methacrylated48 visible photoinitiators, and the final conversion of TMPTA is 39% and 65%, respectively. Therefore, PI-2 may be used as efficient visible photoinitiators for free radical polymerization.
 |
| | Fig. 7 Photopolymerization profiles of TMPTA in laminate upon upon a xenon lamp exposure in the presence of PI-2 (1%, w/w) (1), PI-2/DMA (1%/5%, w/w) (2), PI-2/MDEA (1%/5%, w/w) (3). | |
Conclusions
In summary, 2-allyl(methyl)amino-9H-thioxanthen-9-one (PI-1), 2-benzyl(methyl)amino-9H-thioxanthen-9-one (PI-2) and 2-butyl(methyl)amino-9H-thioxanthen-9-one (PI-3) featured with high absorption and high molar extinction coefficients in the visible range have been prepared and characterized. According to the photophysical and photochemical studies, the hydrogen abstraction/radical transfer processes occur to generate initiating radicals between the triplet PIs and hydrogen donors (the ground state PIs or DMA). Among them, PI-2 has showed best photoinitiating ability evidenced by the fact that it initiates the free radical polymerization of HDDA in the presence and absence of DMA under xenon light exposure (28 mW cm−2) for 0.5 h with 92% and 52% conversion of double bond, respectively. For TMPTA, the conversion of PI-2 and PI-2/MDEA systems is 32% and 62% under xenon lamp exposure for 10 min. Results showed that the introduction of the group which could form carbon radical easily such as benzyl amine could accelerate the photoinitiating procedure.
Acknowledgements
The financial support from the National Science Foundation of China (20844005) is greatly appreciated.
Notes and references
- J. P. Fouassier and J. Lalevée, Photoinitiation, Photopolymerization and Photocuring, Fundamentals and Applications, Hanser Verlag, Munich, Germany, 1995 Search PubMed.
- J. P. Fouassier and J. Lalevée, Photoinitiators for Polymer Synthesis: Scope, Reactivity and Efficiency, Wiley-VCH, Weinheim, 2012 Search PubMed.
- J. P. Fouassier and J. F. Rabek, Radiation Curing in Polymer Science and Technology, Chapman and Hall, London, 1993 Search PubMed.
- J. G. Drobny, Radiation Technology for Polymers, CRC Press, Boca Raton, London, New York, 2nd edn, 2010 Search PubMed.
- J. F. Rabek, Mechanisms of Photophysical Processes and Photochemical Reactions in Polymers: Theory and Applications, Wiley, New York, 1987 Search PubMed.
- J. Hiller, J. D. Mendelsohn and M. F. Rubner, Nat. Mater., 2002, 1, 59–63 CrossRef CAS PubMed.
- N. S. Allen, J. Photochem. Photobiol., A, 1996, 100, 101–107 CrossRef CAS.
- N. Moszner and U. Salz, Macromol. Mater. Eng., 2007, 292, 245–271 CrossRef CAS.
- N. Moszner and U. Salz, Prog. Polym. Sci., 2001, 26, 535–576 CrossRef CAS.
- J. H. Moon, J. Ford and S. Yang, Polym. Adv. Technol., 2006, 17, 83–93 CrossRef CAS.
- J. P. Fouassier and F. Morlet-Savary, Opt. Eng., 1996, 35, 304–312 CrossRef CAS PubMed.
- J. Jakubiak and J. F. Rabek, Polimery, 1999, 44, 447–461 CAS.
- M. A. Tehfe, J. Lalevée, F. M. Savary, B. Graff and J. P. Fouassier, Macromolecules, 2012, 45, 356–361 CrossRef CAS.
- J. Lalevée, F. Dumur, C. R. Mayer, D. Gigmes, G. Nasr, M. A. Tehfe, S. Telitel, F. M. Savary, B. Graff and J. P. Fouassier, Macromolecules, 2012, 45, 4134–4141 CrossRef.
- M. A. Tehfe, F. Dumur, B. Graff, F. M. Savary, J. P. Fouassier, D. Gigmes and J. Lalevée, Macromolecules, 2013, 46, 3761–3770 CrossRef CAS.
- H. Mokbel, F. Dumur, S. Telitel, L. Vidal, P. Xiao, D. L. Versace, M. A. Tehfe, F. M. Savary, B. Graff, J. P. Fouassier, D. Gigmes, J. Toufaily, T. Hamieh and J. Lalevée, Polym. Chem., 2013, 4, 5679–5687 RSC.
- M. V. Encinas, A. M. Rufs, T. Corrales, F. Catalina, C. Peinado, K. Schmith, M. G. Nevmann and N. S. Allen, Polymer, 2002, 43, 3909–3913 CrossRef CAS.
- T. Corrales, F. Catalina, C. Peinado, N. S. Allen, A. M. Rufs, C. Bueno and M. V. Encinas, Polymer, 2002, 43, 4591–4597 CrossRef CAS.
- T. Corrales, F. Catalina, N. S. Allen and C. Peinado, J. Photochem. Photobiol., A, 2005, 169, 95–100 CrossRef CAS PubMed.
- H. Akat, B. Gacal, D. K. Balta, N. Arsu and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2109–2114 CrossRef CAS.
- Y. Wen, X. Jiang, R. Liu and J. Yin, polymer, 2009, 50, 3917–3923 CrossRef CAS PubMed.
- D. K. Balta, N. Arsu, Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2007, 40, 4138–4141 CrossRef CAS.
- D. K. Balta, N. Arsu, Y. Yagci, A. K. Sundaresan, S. Jockusch and N. J. Turro, Macromolecules, 2011, 44, 2531–2535 CrossRef CAS.
- D. K. Balta, G. Temel, G. Goksu, N. Ocal and N. Arsu, Macromolecules, 2012, 45, 119–125 CrossRef CAS.
- M. Aydin, N. Arsu and Y. Yagci, Macromol. Rapid Commun., 2003, 24, 718–723 CrossRef CAS.
- M. Aydin, N. Arsu, Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2005, 38, 4133–4138 CrossRef CAS.
- D. K. Balta, G. Temel, M. Aydin and N. Arsu, Eur. Polym. J., 2010, 46, 1374–1379 CrossRef CAS PubMed.
- G. Yilmaz, S. Beyazit and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1591–1596 CrossRef CAS.
- G. Yilmaz, B. Aydogan, G. Temel, N. Arsu, N. Moszner and Y. Yagci, Macromolecules, 2010, 43, 4520–4526 CrossRef CAS.
- H. Tar, D. S. Esen, M. Aydin, C. Ley, N. Arsu and X. Allonas, Macromolecules, 2013, 46, 3266–3272 CrossRef CAS.
- S. K. Dogruyol, Z. Dogruyol and N. Arsu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4037–4043 CrossRef CAS.
- S. K. Dogruyol, Z. Dogruyol and N. Arsu, J. Lumin., 2013, 18, 98–104 CrossRef PubMed.
- G. Yimlaz, A. Tuzun and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5120–5125 CrossRef.
- N. Karaca, D. K. Balta, N. Ocal and N. Arsu, J. Lumin., 2014, 146, 424–429 CrossRef CAS PubMed.
- D. Tunc and Y. Yagci, Polym. Chem., 2011, 2, 2557–2563 RSC.
- H. Fujiwara and I. Okabayashi, Chem. Pharm. Bull., 1994, 42, 1322–1324 CrossRef CAS.
- Y. Q. Shen, H. D. Tang, Y. H. Zhan, E. A. V. Kirk and W. J. Murdoch, Nanomedicine, 2009, 5, 192–201 CrossRef CAS PubMed.
- M. V. Encinas, A. M. Rufs, T. Corrales, F. Catalina, C. Peinado, K. Schmith, M. G. Neumann and N. S. Allen, Polymer, 2002, 43, 3909–3913 CrossRef CAS.
- T. Tamaki, Bull. Chem. Soc. Jpn., 1979, 52, 1031–1033 CrossRef CAS.
- T. Okada, T. Mori and N. Mataga, Bull. Chem. Soc. Jpn., 1976, 49, 3398–3402 CrossRef CAS.
- D. K. Balta, N. Arsu, Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2007, 40, 4138–4141 CrossRef CAS.
- S. Asmussen and C. Vallo, J. Photochem. Photobiol., A, 2009, 202, 228–234 CrossRef CAS PubMed.
- W. F. Schroeder, S. L. Asmussen, W. D. Cook and C. I. Vallo, Polym. Int., 2011, 60, 1362–1369 CAS.
- T. H. Koch and A. H. Jones, J. Am. Chem. Soc., 1970, 92, 7503–7505 CrossRef CAS.
- R. F. Bartholomew, R. S. Davidson, P. F. Lambeth, J. F. McKellar and P. H. Turner, J. Chem. Soc., Perkin Trans. 1, 1972, 2, 577–582 RSC.
- R. F. Bartholomew and R. S. Davidson, J. Chem. Soc. C, 1971, 2342–2346 RSC.
- R. S. Davidson, P. F. Lambeth and M. Santhanam, J. Chem. Soc., Perkin Trans. 1, 1972, 2, 2351–2355 RSC.
- P. Xiao, F. Dumur, M. Frigoli, M. A. Tehfe, B. Graff, J. P. Fouassier, D. Gigmes and J. Laléeve, Polym. Chem., 2013, 4, 5440–5448 RSC.
- M. A. Tehfe, F. Dumur, B. Graff, F. M. Savary, D. Gigmes, J. P. Fouassier and J. Laléeve, Polym. Chem., 2013, 4, 3866–3875 RSC.
- J. Lalevée, B. Graff, X. Allonas and J. P. Fouassier, J. Phys. Chem. A, 2007, 111, 6991–6998 CrossRef PubMed.
- T. Li, Polym. Bull., 1990, 24, 397–404 CrossRef CAS.
- M. A. Tasdelen, B. Kiskan and Y. Yagci, Macromol. Rapid Commun., 2006, 27, 1539–1544 CrossRef CAS.
- M.-A. Tehfe, F. Dumur, B. Graff, J.-L. Clément, D. Gigmes, F. Mort-Savary, J.-P. Fouassier and J. Lalevée, Macromolecules, 2013, 46, 736–746 CrossRef CAS.
- J. Lalevée, N. Blanchard, M. A. Tehfe, C. Fries, F. Morlet-Savary, D. Gigmes and J. P. Fouassier, Polym. Chem., 2011, 2, 1077–1084 RSC.
- P. Xiao, F. Dumur, B. Graff, J. P. Fouassier, D. Gigmes and J. Lalevée, Macromolecules, 2013, 46, 6744–6750 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 










