
Peptide-decorated gold nanoparticles via strain-promoted azide–alkyne cycloaddition and post assembly deprotection†
Abstract
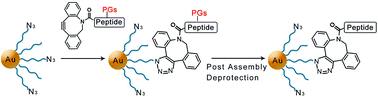
A new method combining an interfacial strain-promoted azide–alkyne cycloaddition and post assembly deprotection (SPAAC-PAD) has been developed for the well-defined functionalization of small, water-soluble gold nanoparticles with oligopeptides.
 Please wait while we load your content...
Please wait while we load your content...

