
Microsol-electrospinning for controlled loading and release of water-soluble drugs in microfibrous membranes
Abstract
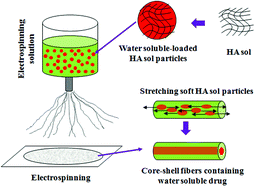
Water-solubility facilitates drug transportation and distribution of drugs throughout the body and hence effectively promotes their absorption. While there have been a number of techniques for incorporating water-soluble drugs into electrospun fibers to realize sustained release of them, problems including burst and uncontrolled release still remain to be solved. In this study, we developed a microsol-electrospinning technique for fabricating core–shell microfibers to achieve incubated, controlled and sustainable release of water-soluble drugs such as chloroquine (CQ). In this approach, nanoparticles made of CQ-loaded hyaluronic acid (HA) sol were first prepared using the emulsification method. Next, the HA-sol nanoparticles were dispersed in poly(L-lactide) (PLLA) electrospinning solution to form a uniform suspension, which was used for fabricating composite microfibers through microsol-electrospinning. Judging from SEM and TEM, the composite microfibers had smooth, uniform morphology and core–shell structure. Further tests showed that the microsol-electrospun microfibers had similar physical, chemical, and mechanical properties as microfibers fabricated using a conventional electrospinning approach. In vitro drug release tests showed that compared to conventional electrospun microfibers, the burst release of CQ was significantly reduced in microsol-electrospun microfibers. Meanwhile, the release time of CQ was markedly extended, being as long as more than 40 days. Importantly, the drug release rate could be readily adjusted by changing the concentration of microsol particles and the amount of drug in the microfibers. Together, findings from this study have revealed that microsol-electrospinning is a facile technique for loading water-soluble drugs into electrospun microfibers and releasing them in a controlled fashion, which may expand the applications of water-soluble drugs.
 Please wait while we load your content...
Please wait while we load your content...

